Electronic ISSN 2287-0237
Thalassemia and sickle cell disease (SCD) are the two most widely distributed inherited hemoglobin-opathies in the world. Thalassemias are prevalent in Mediterranean, Middle Eastern, South and Southeast Asian countries, while sickle cell diseases originate from countries across Africa; both have become widespread globally due to modern migration patterns.1
In beta-thalassemia disease, pathogenesis involves absent or impaired synthesis of the beta-globin chains which constitute adult hemoglobin molecules. This genetic defect causes ineffective erythropoiesis through the whole process of proliferation and maturation of the erythroid precursors, and severe apoptosis is evident due to an accumulation of excess alpha-2 chains. Hemoglobin E is abnormal hemoglobin, formed by a single nucleotide substitution: lysine replacing glutamic acid at the 26th position of the beta-globin chain of hemoglobin A.
In SCD, a single nucleotide substitution occurs: valine replaces glutamic acid at the 6th position of the beta-globin chain of hemoglobin A, forming the pathological hemo- globin called hemoglobin S. The abnormality of hemoglobin S polymerization causes an alteration of the red blood cell structure to a stable sickle shape. Consequently, episodic anemia and acute and chronic vaso-occlusion of small and large vessels in many organs occur and lead to polymorphic clinical features of the disease. Intramedullary apoptosis is also present but is generally milder than that seen in thalassemia.1
Many thalassemic patients need continuous red blood cell replacement through a regular red blood cell transfusion program. Without adequate transfusions, thalassemic patients have remarkable skeleton deformities due to intramedullary expansion, as well as hepatomegaly and splenomegaly due to the proliferation of the hematopoietic system with extramedullary hematopoiesis. Since both thalassemia and SCD are genetic diseases in which the genetic defect is expressed in the hematopoietic system, they are both considered to be curable by allogeneic cellular gene therapy through hematopoietic the cell transplan- tation (HCT) method.1
The basic rationale of allogeneic HCT in thalassemia consists of substituting thalassemic stem cells bearing ineffective erythropoiesis with healthy allogeneic stem cells capable of effective erythropoiesis.2,3 This cellular replacement therapy is not limited to only the diseased erythropoietic component, but also to the replacement of the entire hematopoietic system. Nevertheless, the ultimate goal is to obtain a long-lasting, possibly permanent, effective correction of chronic hemolytic anemia, subsequently avoiding any further transfusion requirements and preventing associated complications such as iron overload and major organ damage, etc.
Transfusion-dependent beta-thalassemia and alpha- thalassemia are highly prevalent hereditary hematologic disorders in Thailand.4 As mentioned earlier, hematopoietic stem cell transplantation is the only curative treatment of these diseases. Most of the reports worldwide are of bone marrow transplantation.5 Increasing recently umbilical cord blood transplantations (CBT) have been performed in a variety of malignant and non-malignant diseases, including thalassemia.4,6-8
In this study we assessed the outcomes of thalassemia and hemoglobinopathy patients undergoing allogeneic HCT in our medical center. Not only the prospects of engraftment, but our analysis also focused on the occurrence of potential complications such as acute and chronic GvHD, treatment related morbidity and mortality, overall survival (OS) and disease-free survival (DFS).
All transfusion-dependent thalassemia and hemo- globinopathy patients undergoing allogeneic HCT from February 2009 through December 2013 were included in this case series study. The transplant procedures were performed at the Bangkok Blood and Marrow Stem Cell Transplantation Unit, Wattanosoth Hospital, Bangkok Hospital Group, Bangkok, Thailand. Donors were selected from HLA-identical siblings of each patient by HLA typing methods. Parents of the patients and donors signed informed consent before treatment and donation. Likewise, parents of cord blood newborn donors also signed informed consent prior to cord blood collection and cryopreservation.
Our hematopoietic cell transplantation procedures complied with standard practice and considerations. Bone marrow was considered as a first-choice source of hema- topoietic cells from the existing donors, HLA-identical siblings, whereas umbilical cord blood was considered from newborn siblings if applicable. Every recipient (patient) was implanted with a double-lumen central venous catheter, namely Hickman, by a vascular surgeon, prior to starting the pre-transplant medication. We used a myeloablative conditioning regimen approach for the patients who were pre-classified as Pesaro class I or II. Pre-transplant conditioning chemotherapy was composed of busulfan9,10 (Bu) 4-4.8mg/m2/day for 4 days and fludarabine (Flu) 35mg/m2/day for 5 days and rabbit anti-thymocyte globulin (rATG) 1, 2, 3mg/kg/day for 3 consecutive days for thalassemia patients, while Bu (same dose) and cyclophosphamide (Cy) 50mg/kg/day for 4 days and rATG (same dose) were used for sickle cell disease patients. This preparatory regimen usually took about 8 days, and was completed at least 1 day before infusion day (transplant day = day zero).
On transplant day for bone marrow transplantation (BMT) cases, bone marrow stem cells were obtained from the donor by a bone marrow harvest under general anesthesia in the operating room. Each fresh bone marrow product was subsequently infused into the recipient’s blood circulation via Hickman catheter on the same day. If there was major blood group ABO incompatibility between donor and recipient, the donor’s bone marrow product had to undergo a red blood cell depletion process before infusion. In contrast, if a minor ABO incompatibility was identified, a plasma removal process of the donor’s marrow product was performed before stem cell infusion.
In the case of umbilical cord blood transplant (CBT), umbilical cord blood units that had been identified were transported to the transplant unit under frozen conditions by a portable cryo-tank and thawed just before infusion into the recipient’s great vein through Hickman catheter. In the case of combined cord blood and bone marrow transplant (CB+BMT), the standard cord blood infusion procedure was performed and followed soon after by a fresh marrow infusion. The CD34+ cell dose per kilogram of the recipient’s body weight was considered an essential marker to determine the hematopoietic cell dose.
During the pre- and early post-transplant period, prophylactic antimicrobial medication was administered. Protective environment precautions were applied. The recipient remained in an isolated, positive-pressure, high efficient particulate aerosol (HEPA) filtration room. All packed red cell units and platelet-pheresis units were filtered and irradiated before being transfused as a supportive treatment.
After stem cell infusion, graft-versus-host disease (GvHD) prophylaxis was administered, consisting of intravenous cyclosporine and short-course, low-dose methotrexate injection for BMT recipients. GvHD prophylaxis consisted of only cyclosporine for CBT recipients. All patients were given recombinant human granulocyte colony-stimulating factor (G-CSF) 5 microgram/kg after transplantation. It was infused over a ten minute period intravenously starting from 4 hours after HCT and continuing once daily until neutrophil recovery exceeded 2 x 109/l for 2 consecutive days.
Meanwhile, neutrophil recovery or engraftment was defined as the first day of an absolute neutrophil count (ANC) over 0.5 x 109/l for 3 consecutive days. Platelet recovery was defined as the first day of a platelet count over 20 x 109/l for 3 consecutive days without platelet transfusion support. Graft failure or rejection was defined if ANC did not rise, or no neutrophil engraftment within 28 days after HCT or ANC declined below 0.2 x 109/l after initial recovery. After neutrophil engraftment, acute GvHD would be diagnosed and graded according to previously reported criteria.11,12 Recipients living 100 days post-transplant with sustained donor engraftment were considered to be evaluated for chronic GvHD, as described.13
The CMV DNA viral load was monitored weekly in all post-transplant patients when white blood cells had recovered, in order to detect any CMV activity. Patients who had reactivation of CMV were treated with pre-emptive therapy of ganciclovir at a conventional dosage and they were monitored until the viral replication became undetected.14
There were fifteen thalassemia and hemoglobinopathy patients with ages ranging from 2 to 15 years 11 months old (median 5 years), all undergoing allogeneic HLA- matched sibling donor HCT. The details of patient char- acteristics are reported in Table 1. Thirteen patients were diagnosed with transfusion-dependent beta-thalassemia/ hemoglobin E diseases, one was diagnosed with trans- fusion-dependent alpha-thalassemia disease (hemoglobin H Constant Spring = alpha-thalassemia 1/alpha Constant Spring), and one was diagnosed with sickle cell disease with frequently recurrent painful vaso-occlusive crises. The patients were 12 males and 3 females. Their nationalities consisted of 11 Thais, 1 French-Thai, 1 Bangladeshi, 1 Lao, and 1 Omani. The patients’ body weight ranged from 11.1kg to 50kg (median 17.3kg). The fourteen thalassemia patients were classified based on Pesaro classification into class I (n=10), class II (n=4). There were no class III patients in our series.
Table 1: Clinical characteristics of enrolled patients. (n = 15).
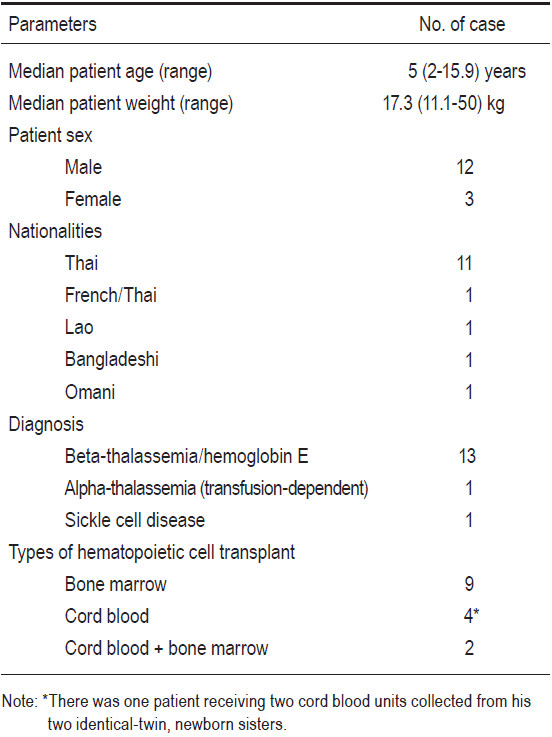
There were 16 related donors overall who were fully- HLA-matched with their corresponding 15 recipients. (This is because there were two identical-twin, newborn sisters whose two cord blood units were collected and later used for transplantation to one recipient, their elder brother). With regards to the status of the donors, 5 had normal typing, 6 had beta-thalassemia traits, 3 had hemoglobin E traits, 1 had alpha-thalassemia one traits, and 1 had sickle cell traits (see Table 2 and 3). The source of the donors’ hematopoietic cells consisted of 9 bone marrow (BM), 5 umbilical cord blood (CB) units, and 2 cord blood plus bone marrow (CB+BM). BM donors were 6 males and 5 females, with ages ranging from 2 to 18 years old (median 4 years) (see Table 2). Five CB units were collected from 5 term neonates when they were delivered, two of whom were identical twins. There were two 2-year-old male donors who donated their cord blood units and bone marrow for their respective elder brothers who had thalassemia diseases.
Among the 9 patients to receive a transplant of BM, the median CD34+ cell dose was 12.3 (range 5.6 to 34.7) x 106/kg body weight. Among the 4 patients to receive a transplant of CB stem cells, the median CD34+ cell dose was 2.3 (range 1.6 to 3) x 105/kg body weight.
The probability of initial hematopoietic recovery by day 30 was equal to 100% (15 out of 15 patients). Neutrophil recoveries were evident on days +10 to +23 (median day +14), and platelet recoveries were observed on days +19 to +64 (median day +40).
Engraftment evaluations were studied by chimerism analysis using the microsatellite, short tandem repeat method. For cases of gender mismatch between recipients and donors, the techniques of fluorescent in-situ hybrid- ization (FISH) of XX-XY chromosome were also used. In terms of engraftment, no patients experienced graft failure. Every patient was neutrophil engrafted. Twelve patients achieved complete donor engraftment. Two patients from the BMT group and one from the CBT group developed stable mixed-chimerism states with donor predominance and they were no longer blood transfusion-dependent, nor required further transfusion. The summaries are reported in Table 4.
Every recipient and donor had Rh positive blood group. Eleven recipients had same blood group ABO as their corresponding donors. There were three recipients of major blood group B-O incompatibility and their blood groups were altered from pre-HCT “O” to post-HCT “B”. On the other hand, there was one case of minor blood group A-AB incompatibility and this patient’s blood group was switched from pre-HCT “AB” to post-HCT “A” (see Table 5).
Table 2: Clinical characteristic of bone marrow donors [bone marrow transplant (BMT) & cord blood and marrow transplantation(CB+BMT)] (n = 11).
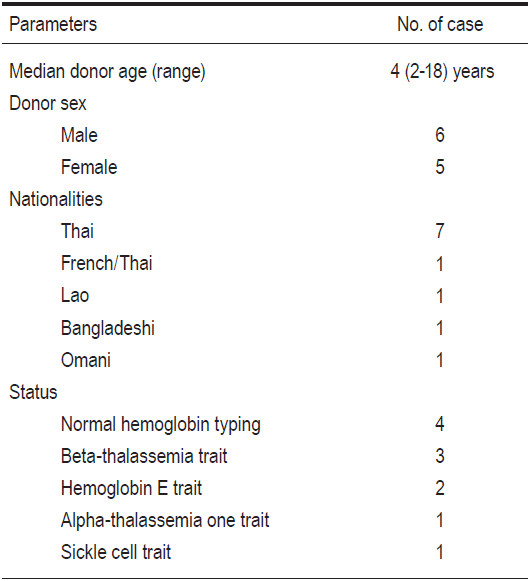
Table 3: Clinical characteristic of isolated cord blood newborn donors.

Table 4: Outcomes of recipients after allogeneic HCT (n=15).
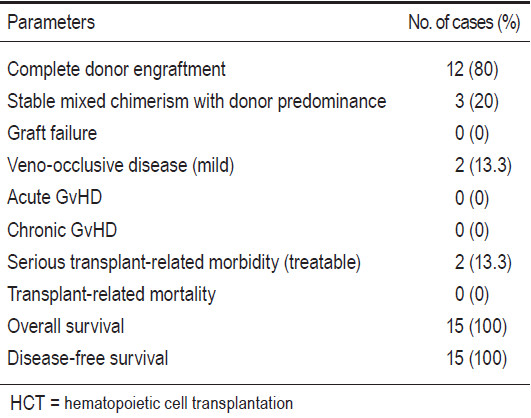
Table 5: Alteration of blood group in ABO-mismatched recipients after allogeneic HCT.

No patients developed acute or chronic GvHD. The overall probability of mortality was equal to 0%. Two patients had mild veno-occlusive diseases of the liver which were later completely reversible by means of conservative treatment. There were no cases of mortalities, but some serious morbidity due to infectious complications occurred. One patient had severe but treatable pneumocystis pneumonia at 4 months post CBT. Another patient had catheter-related gram negative bacilli septicemia at 2 months post CB+BMT and recovered after receiving intravenous antibiotics after the Hickman central venous catheter removal. Both patients remain well to date. Moreover, there were two recipients who developed CMV reactivation, with no clinical manifestation, during their respective 3 and 4 months post HCT. They responded well with pre-emptive ganciclovir therapy and were able to discontinue the medicine within 2 months.
The median follow-up time for all 15 patients was 2 years 2 months (1 month to 4 years 10 months). Overall (OS) and disease-free survival (DFS) rates were 100% and 100% for all patients (n=15). Based on the Pesaro risk class, the OS and DFS for class I thalassemia patients (n=10) were 100% and 100%, and class II patients (n=4) were 100% and 100%, respectively (see Table 4).
These globally widespread single-gene disorders: beta-thalassemia and sickle cell disease (SCD), can only be cured by means of allogeneic hematopoietic cell trans- plantation (HCT). Although improvements in conservative treatment have considerably improved the prognosis of non-HCT, transfusion-dependent thalassemia cases, disease- and treatment-related complications in these patients progress over time, causing inevitable morbidity and eventual shortened life expectancy.17 HCT still remains the only cure currently available for this group of patients.
Treatment outcomes by HCT for thalassemia have substantially improved over the past two decades, with the development of updated pre-transplant preparative regimens, advancement in preventive strategies, and control of transplant-related complications.17 Outcomes nowadays are much improved compared with the 1980s and 1990s, with more than 90% of patients surviving
transplantation and more than 80% of them being disease- free in several medical centers worldwide.2,15,16 Some studies revealed a risk class-based transplantation approach led to disease-free survival probabilities of 90%, 84%, and 78% for class I, II, and III thalassemia patients, respectively.1 Some revealed a risk class-based approach to transplantation in thalassemia led to a disease-free survival probability of 87%, 85% and 82% in classes I, II and III patients, respectively. Adult thalassemia patients are higher risk patients for transplant-related toxicity due to an advanced phase of disease and have a cure rate of 62% with current treatment protocols.17
Based on our study results, the OS and DFS were excellent with 100% survival in both categories. However, the numbers of cases is small, only 15. We will have to gather more case enrollments and repeat the analysis in the future. Favorable factors are summarized as follows:1)There were only class I and II patients,no class III, 2) Every patient underwent HLA-identical sibling HCT, 3) Adequate hematopoietic stem cell doses had been estimated before the actual transplantation process (CBT) started or were obtained by bone marrow harvest before stem cells infusion (BMT), 4) Peripheral blood was not used as a source of stem cells since there were several reports of enhancing incidence of GvHD after peripheral blood stem cell transplantation, 5) Our current conditioning regimen, GvHD prophylaxis, and antimicrobial prevention was effective, 6) Our transplant care team cared for the patients safely throughout pre-, during, and post-transplant periods and forestalled potentially fatal complications.
Although allogeneic HCT has such a high success rate, the major limitation to whether the patient is eligible for HCT is the lack of an HLA-identical sibling donor for most affected patients. In fact, only 25-30% of thalassemia patients have a matched sibling donor available. Therefore, there is a need to develop alternative sources of stem cell donations. To date high-resolution HLA typing tests have enabled some patients to have the chance to undergo a transplant from an HLA-matched unrelated volunteer donor, with results comparable with those undergoing an HLA-identical sibling transplantation.17
The near-future prospects of our HCT team include: dealing with good prognostic-factor patients or HLA- identical sibling donors, and at risk class III patients, and undertaking HLA-matched unrelated donor searching and procurement. The HCT team needs to keep updating its knowledge base and keep reviewing reports and experiences from other modern medical practices. This is crucial to keep seeing good treatment results with future patients.
Our experience in HCT for thalassemias and hemo- globinopathies has been very favorable. The HLA- matched related donor HCT has had the best success rate. Cord blood transplantation yielded the same excellent outcomes as bone marrow transplantation. Nonetheless, we plan to regularly follow up with post-transplant patients to detect any possible complications and to determine long-term outcomes.
Acknowledgements
The author wishes to thank Kornkiat Vongpaisarnsin, MD, Department of Forensic Medicine, Faculty of Medicine, Chulalongkorn Universitiy for evaluating the chimerism analysis, and Verayuth Praphanphoj, MD, Rajanukul Institute for assessing the fluorescent in-situ hybridization (FISH) of XX-XY chromosome in gender- mismatch cases. The author also would like to thank Kamol Ruengthong, MD; Tavee Surojnametakul, MD; Karn Danpongchareon, MD; Ananchai Tatiyaapitham, MD; Porntep Suandork, MD; Darin Aranwutikul, MD; Thitirat Ratanasila, MD; Supara Leechasan, MD; Sirimon Piyavunno, MD; all other Pediatric consultants; the nursing staff of the Stem Cell Transplant Unit, in-patient wards 6W, 14D, Pediatric out-patient clinic, operating room, and all Blood Bank and laboratory staff.









