Electronic ISSN 2287-0237
To assess outcomes of hematopoietic cell transplantation (HCT) for thalassemias and hemoglobinopathies in a single medical center in Thailand.
Case series study for thalassemia and hemoglobinopathy patients undergoing HCT at Bangkok Hospital Medical Center (BMC) from February 2009 through December 2013.
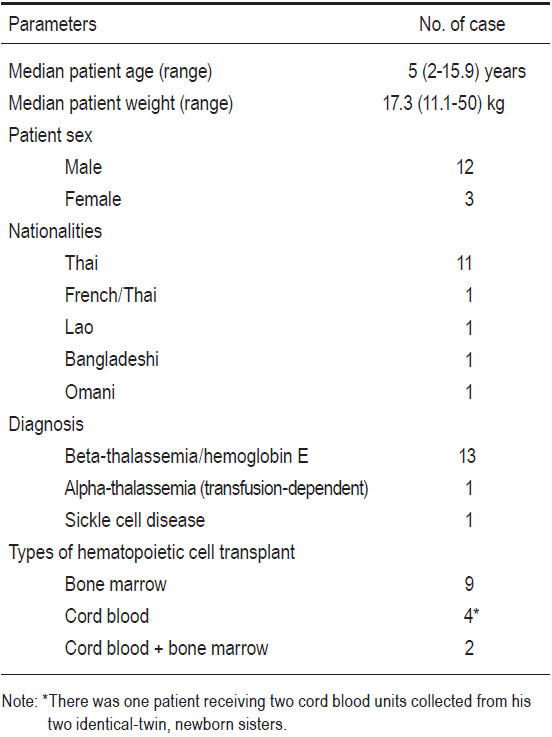
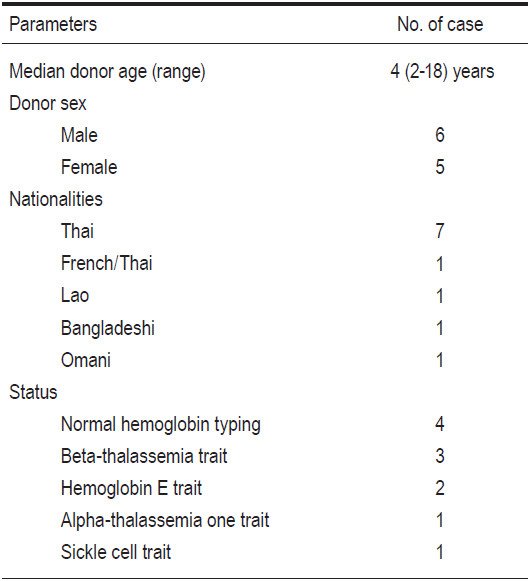
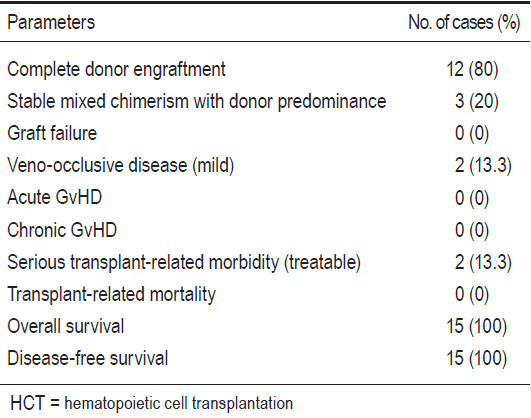
There were a total of 15 patients. Eleven cases were Thais, 1 was French-Thai, 1 was Bangladeshi, 1 was Lao, and 1 patient was Omani who had sickle cell disease (SCD). Thirteen cases were diagnosed as beta-thalassemia/hemoglobin E diseases, 1 as a transfusion-dependent alpha-thalassemia, and 1 as SCD. Among the 15 HCTs, 9 patients underwent bone marrow transplant (BMT), 4 patients underwent umbilical cord blood transplant (CBT), and 2 patients underwent combined cord blood and marrow transplantation (CB+BMT). All donors were related and fully-HLA-matched. The male to female patient ratio was 12:3. The patients’ ages at transplant varied from 2 years to 15 years 11 months with a median of 5 years. The patient’s body weight varied from 11.1 kilogram (kg) to 50kg (a median of 17.3kg). According to the Pesaro classification, among the 14 thalassemia patients there were 10 class I, and 4 class II patients. Busulfan, fludarabine, and rabbit ATG were mainly used as the myeloablative conditioning regimen. Cyclosporine and short-course methotrexate were mainly used as graft-versus-host disease (GvHD) prophylaxis in BMT and CB+BMT groups, while cyclosporine alone was used in the CBT group. CD34+ cell doses per kilogram body weight recipients ranged from 5.6 to 34.7x10^6 (a median of 12.3x10^6) in the BMT group (n=9), and from 1.6 to 3x10^5 (median 2.3x10^5) in the CBT group (n=4). Complete donor engraftments were achieved in 12 patients. Mixed-chimerism states with donor predominance were present in 2 patients from the BMT group and 1 patient from the CBT group. No patients experienced graft failure. Neutrophil recoveries were evident on days +10 to +23 (median day +14), and platelet recoveries were observed on days +19 to +64 (median day +40). No patients developed acute or chronic GvHD. There were no mortalities. Median follow-up time for all patients was 2 years 2 months (1 month to 4 years 10 months). Overall (OS) and disease-free survival (DFS) were 100% and 100% for all patients (n=15). Based on the risk class, the OS and DFS for class I thalassemia patients (n=10) were 100% and 100%, and class II patients (n=4) were 100% and 100%, respectively.
Our experience in HCT for thalassemias and hemoglobinopathies has been very favorable. HLA-matched related donor HCT yielded the best success rate. Anyhow, regular follow-up visits are encouraged to detect any possible complications and to determine long term outcomes.
hematopoietic cell transplantation, thalassemia, sickle cell disease
10.31524/bkkmedj.2014.02.001