Electronic ISSN 2287-0237
Gastaut type idiopathic childhood occipital epilepsy (G-ICOE) is a rare epileptic syndrome which is considered a form of idiopathic focal epilepsies in childhood. The diagnosis of G-ICOE can be made by particular clinical features together with characteristic electroencephalographic activities (electro- clinical syndrome). In this paper, we present a case of G-ICOE with classic EEG findings and a literature review.
A 17-year-old young gentleman from Bangladesh presented to the Neuroscience Center of Bangkok Hospital for evaluation of epilepsy. The patient had his first seizure when he was seven years old. All of his seizures were stereotypical. Most seizures occurred while he was asleep. The patient was not aware of the seizure and could not recall the entire event. According to witnesses, he was noted to be convulsing all over. His eyes were wide open and rolling upward. The average duration of seizure was one to two minutes. There was no history of tongue biting, urinary/fecal incontinence, or seizure-related injuries.
There was one episode of seizure that occurred during the day. At that time he was traveling with his family on the train, and had forgotten to take his anti- epileptic medication for several days. Prior to the convulsion, he reported that he was feeling drowsy with some changes in his vision which he could not remember exactly, probably isolated white spots in both eyes.
He had undergone evaluations in Bangladesh and Singapore. MRI of the brain was reportedly unremarkable. Prior EEGs revealed epileptiform discharges over the right posterior temporal-occipital regions. He was started on valproic acid, the dose of which was adjusted periodically according to his body weight, and he has been seizure-free over the past four to five years.
His pregnancy and delivery were unremarkable. The patient had a normal growth and development. He was currently doing well in high school. There was no history of febrile convulsion, brain tumor, infection of the central nervous system, stroke, or traumatic brain injury. There was no family history of epilepsy.
On examination, the patient was alert, awake, and oriented to time, place, and person. Detailed ophthalmologic evaluation was within normal limits. There was no evidence of visual field defect. Otherwise his general and neurological examinations were unremarkable.
The patient underwent electroencephalography (EEG) which revealed a normal posterior background of 10-11 Hz. There were abundant sharp and slow wave complexes over the left occipital region, which disappeared when the patient opened his eyes and reappeared with eye closure and during sleep. The patient was responsive, and able to communicate normally when he was awake throughout the EEG recording. (Figure.1-3)
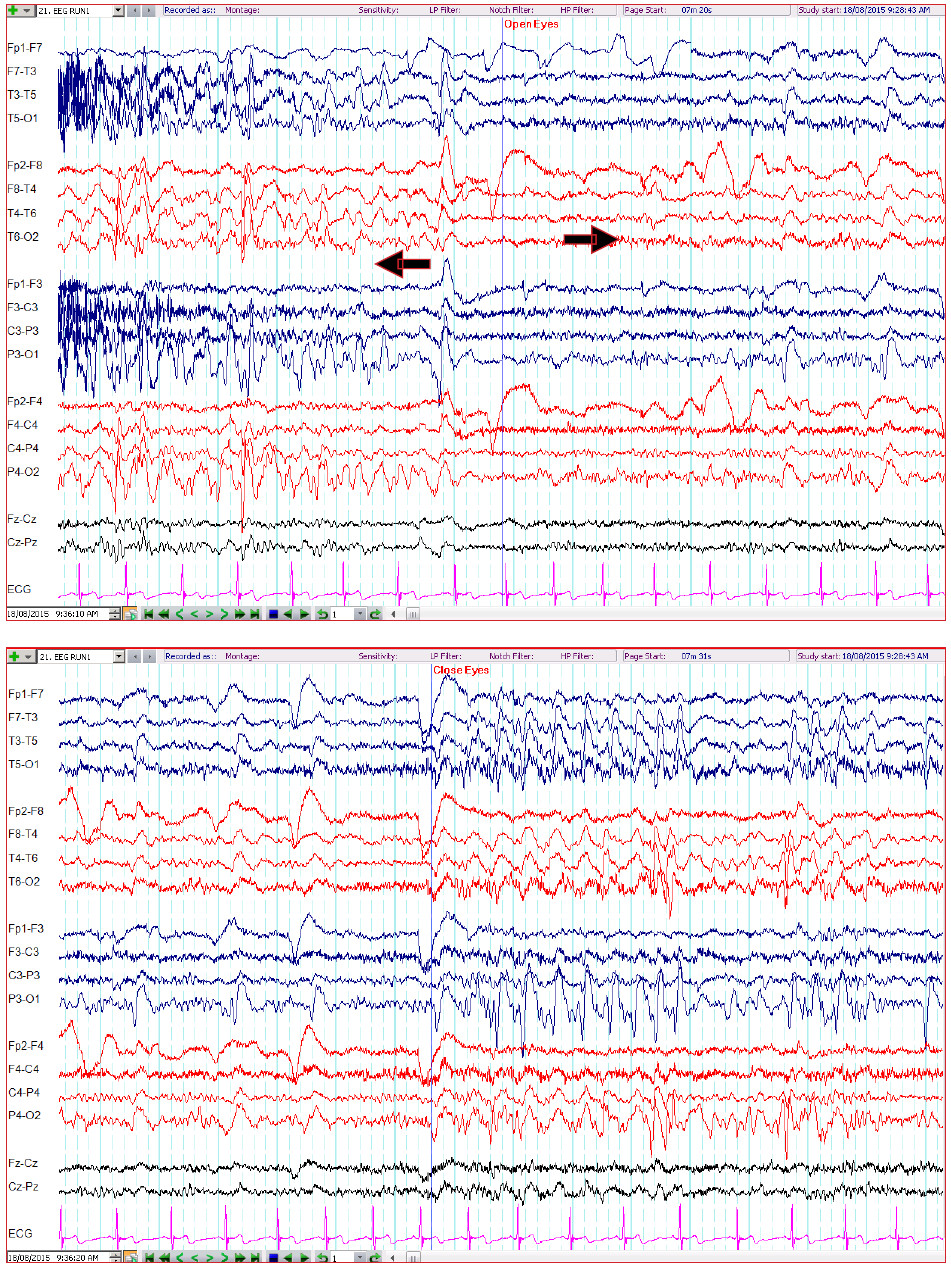
Figure 1-2: The EEG reveal runs of repetitive, high amplitude sharp and slow wave complexes in the occipital regions which appear immediately when the eyes are closed and persist for as long as the eyes remain closed. These epileptiform activities suddenly disappear after the eyes are open. (10-20 electrodesystem; double banana montage; high-pass filter 1 Hz; low-pass filter 70 Hz, notch filter 50 Hz, sensitivity 7 µV)
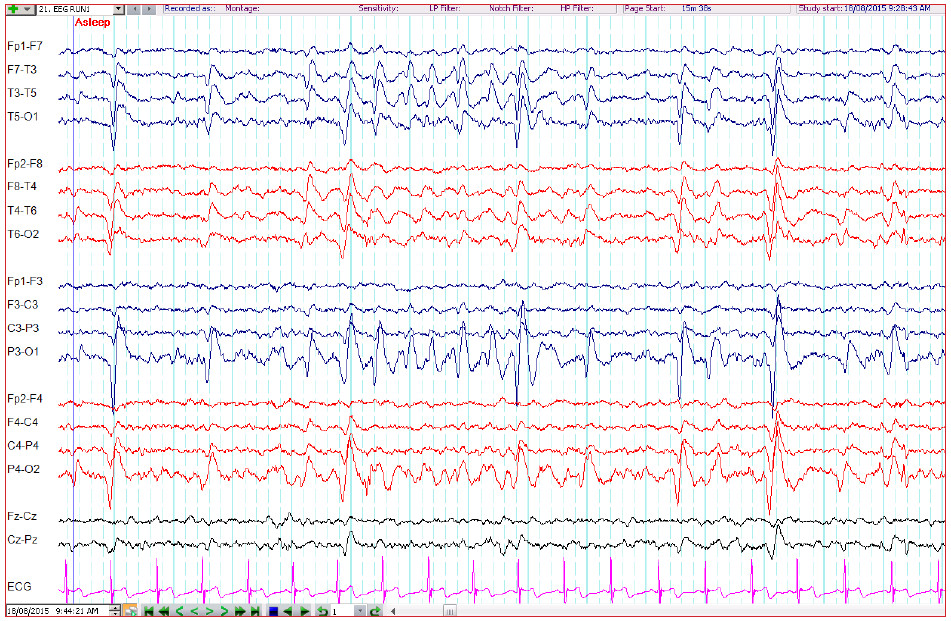 Figure 3: The EEG depicts the run of occipital sharp and slow wave complexes during sleep. (10-20 electrode system; double banana montage; high-pass filter 1 Hz; low-pass filter 70 Hz, notch filter 50 Hz, sensitivity 7 µV)
Figure 3: The EEG depicts the run of occipital sharp and slow wave complexes during sleep. (10-20 electrode system; double banana montage; high-pass filter 1 Hz; low-pass filter 70 Hz, notch filter 50 Hz, sensitivity 7 µV)
The EEG findings of our patient, i.e., runs of occipital sharp and slow wave complexes which attenuate on eye opening or occipital paroxysms, are diagnostic of idiopathic childhood occipital epilepsy. Idiopathic child- hood occipital epilepsy can be classified into two discrete categories of early (Panayiotopoulos syndrome) and late onset (Gastaut type). The classical presentation of each syndrome is quite distinctive. Panayiotopoulos syndrome is notable for its infrequent autonomic seizures that on some occasions can be prolonged (autonomic status epilepticus), predominantly nocturnal, with the usual age of onset between three to five years.1 On the contrary, Gastaut type-idiopathic childhood occipital epilepsy (G-ICOE) generally manifests with frequent, mostly diurnal, brief elementary visual aura or ictal blindness, with the mean onset age of eight years.2 Oftentimes clinical features of each syndrome can be overlapped,3 and both types may be associated with hemi-clonic or generalized convulsion with impairment of consciousness.4
Our patient was diagnosed with Gastaut type idiopathic childhood occipital epilepsy mainly based on his age of onset, the duration of seizure, together with the characteristic EEG findings and the history of rare visual aura. In addition, the lack of autonomic features such as vomiting, pallor, or sweating argues against the diagnosis of Panayiotopoulos syndrome. There are some atypical features in this case, including the tendency of the seizure to be nocturnal and relatively infrequent seizure activities, which can potentially result from concurrent antiepileptic therapy.
Since most patients with G-ICOE suffer from frequent seizures, medical therapy is often mandatory. Carbamazepine has been used with excellent responses,5 but valproic acid, oxcarbazepine, levetiracetam, or other antiepileptic medi- cations are believed to be equally effective.6 The prognosis of G-ICOE is not clear, as there is high possibility of several case series being contaminated by symptomatic occipital epilepsy. Seizure remission occurs in 50-60% of patients after 2-4 years from first onset, and in general the response to antiepileptic drugs is fairly good.6
Gastaut type idiopathic childhood occipital epilepsy (G-ICOE) is a rare epileptic syndrome with the mean age of onset around eight years. Typical clinical features are frequent, brief, elementary visual auras which tend to be diurnal, but the presentation can be overlapped and may be a continuum with its counterpart Panayiotopoulos syndrome. The characteristic EEG findings include occipital paroxysms. Antiepileptic medication is often necessary and prognosis is generally good with appropriate treatment.









