Electronic ISSN 2287-0237
A 62-year-old Ethiopian woman with a 10 year history of hypertension and cirrhosis of unknown cause. Sjogren’s syndrome and systemic lupus erythematosus (discoid lupus, autoimmune hemolytic anemia, positive ANA/ SSA/ antiribosome P/R052) had been diagnosed in 2010. She has been on prednisone 10mg per day, hydroxychloroquine 200 mg per day, candesartan 4 mg per day and Ferli-6, 1 tab three times daily. She denied new rashes, joint stiffness or fever. She had a dry mouth. At her visit she was found to have more severe anemia and and increase of proteinuria. There was no family history of rheumatoid arthritis, SLE or other autoimmune disease.
Physical examination (PE) revealed a normotensive, overweight woman with 1+ ankle edema. Laboratory investigation revealed hemoglobin 7.7 mg/dl, normal com- plement level, negative coomb’s test, albumin 2.0 mg/dl, normal TSH,negative hepatitis virus A and C, proteinuria (1.6 gm/day), creatinine 1.9 mg/dl. The CRP level was 5.03 mg/dl. Chest radiograph was normal.
Bone marrow biopsy revealed normocellular marrow without excess blasts or dysplastic change. A renal biopsy was performed.
Light microscopy findings (Figure 1, 2): The biopsy is a sample of two cores of renal cortex and medulla with 12 glomeruli per section level. All glomeruli reveal variable stages of accumulation of eosinophilic extracellular deli- cately fibrillar material in the mesangium, arteriolar walls and interstitial tissue. There is extensive tubular atrophy with segmentally dense interstitial inflammatory infiltrates consisting of lymphocytes,plasma cells,and mononuclear leukocytes. Some nonatrophic tubules show tubulitis with amorphous material in the lumens.
Immunofluorescence microscopy (IF) findings (Figure 3): six (6) glomeruli are present in each frozen section. Two (2) glomeruli are globally sclerosed. Four (4) glom- eruli show nodular deposition of C3 in some mesangial areas. There is no deposition of IgG, IgM, IgA, Ciq, fibrin, kappa L. C or lambda L. C.
Immunohistochemical (IHC) staining (Figure 4, 5) for amyloid A revealed irregular distribution of amyloid A in mesangial areas, along the tubular basement membrane, in arteriolar walls and interstitial tissue.
Ultrastructural findings (Figure 6): The kidney tissue was obtained from the paraffin block and reprocessing for transmission electron microscope (TEM) study. Randomly arranged non-branching fibrils (8 - 12nm), compatible with amyloid fibrils are seen in the arteriolar wall and interstitium, corresponding with the amorphous material under light microscope. The diagnosis of AA amyloidosis with renal involvement secondary to SLE was established. The patient received colchicine for 1 month then she no longer came in for follow-up visits.
AA amyloidosis is known to be secondary to chronic infectious diseases including tuberculosis, bronchiectasis, chronic osteomyelitis, and chronic inflammatory diseases such as Crohn’s disease, rheumatoid arthritis, ankylosing spondilitis and has been reported in familial Mediterra- nean fever.1 A small number of cases of AA amyloidosis associated with systemic lupus erythematosus (SLE) are reported in the literature. Some previous reports state that the link between AA amyloidosis and SLE might be explained by the presence of additional conditions known to cause amyloidosis. The type of amyloid protein involved has not been clearly established in most of the reported cases.
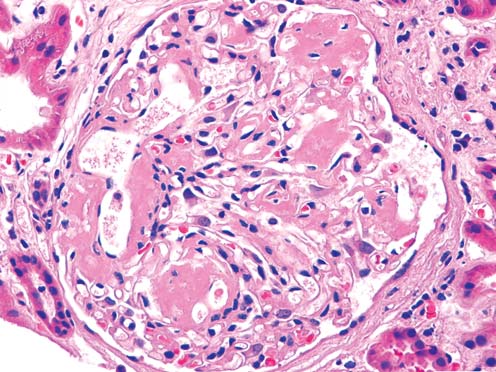
Figure 1: Glomerular deposition of eosinophilic fibrillar material (H&Ex200).
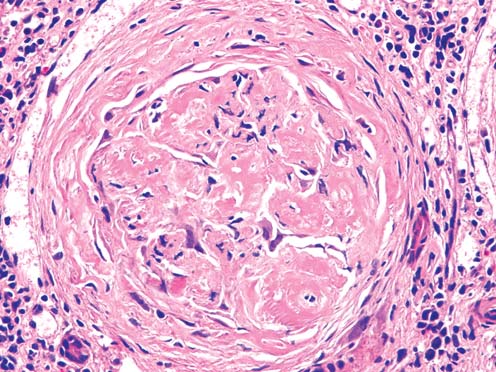
Figure 2: Glomerular deposition of eosinophilic fibrillar material in glomerular structure and periglomerular fibrotic tissue (H&Ex200).
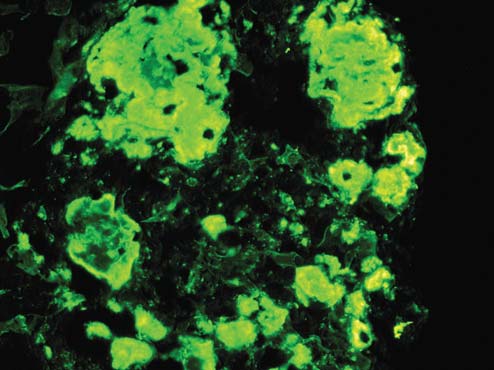
Figure 3: Deposition of C3 in glomerulus (IFx200).
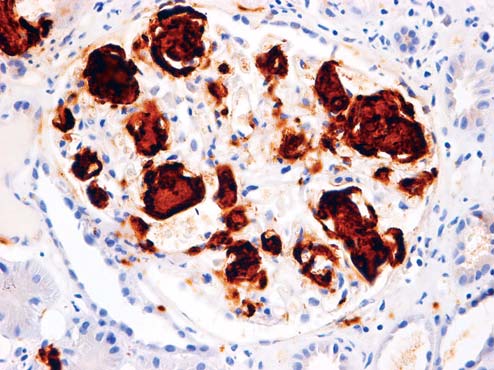
Figure 4: Deposition of amyloid A in glomerulus (IHCx200).
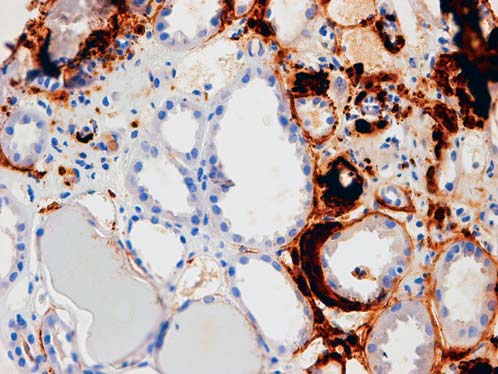
Figure 5: Deposition of amyloid A in peritubular and inter- stitium (IHCx200).
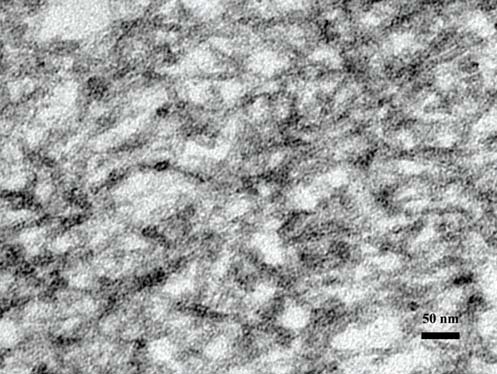
Figure 6: Electron microcopy demonstrating deposition of non-branching 8-12 nm typical of amyloid fibrils in arterio- lar wall (TEMx40,000).
The kidney is the most common site of amyloid fibril deposition with progressive deterioration in renal function.2 Deposition of amyloid fibrils in other organs in patients with SLE such as heart,3 gastrointestinal tract,4 and skin5 have been reported.
Our patient developed two diseases of the immune system, SLE and Sjogren syndrome at the same time. An extensive evaluation of clinical history and labora- tory tests did not reveal any of those abovementioned causes for the development of secondary amyloidosis. Both diseases were clinically inactive, yet the C-reactive protein was high. Amyloidosis usually develops in patients with long-standing SLE as in our case. Occasionally, two diseases have been discovered at the same time.6
The diagnosis of amyloidosis requires a histologic demonstration of amyloid deposits. This is accomplished by staining with Congo red dye and the presence of characteristic fibrils identified by electron microscopy examination. Types of amyloid are indistinguishable by light or electron microscopy. The definitive method used is immunofluorescence or immunohistochemical staining of tissue with antibodies that are directed against known amyloidogenic proteins.
Renal pathology in previous reported cases revealed light microscopy change, compatible only with amyloidosis without immune deposition by immunofluorescence microscopy. Our case showed also glomerular deposition of C3 in high intensity together with amyloid fibrils with no deposits of gamma globulins. The prognostic factor affecting progression to deterioration of renal function is the concentration of the amyloidogenic precursor SAA.7
The amyloidogenic precursor SAA was not tested in our case. The relationship between amyloid fibrils and immunoglobulins or complement components is not well understood. More extensive investigation into these kinetic alterations of immune complexes and AA protein is required.









