Electronic ISSN 2287-0237
The coagulation system is essential for survival. The mainphysiological function of the coagulation system is hemostasis,the sealing of leaks in the vasculature to prevent excessive bloodloss. Tissue factor surrounding the blood vessels activates the ‘classical’or ‘extrinsic’ activation of coagulation. Activation of coagulation triggersa complex and highly regulated cascade system of sequentiallyactivated proteases, culminating in the generation of thrombin (factorIIa), which converts soluble fibrinogen into an insoluble fibrin network,and is a potent activator of blood platelets. Together, activated plateletsand fibrin seal damaged blood vessels (Figure 1).
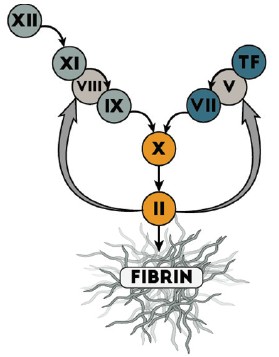
Figure 1: Coagulation can be activated through different pathways.First, through the intrinsic pathway (factor XII and factor XIactivation), triggered by contact with foreign bodies or byinflammatory processes, and secondly as a result of tissue factor(TF) release at sites of vessel damage. Activation of factor X tofactor Xa is the start of the common pathway, which leads toactivation of prothrombin (II) to thrombin (IIa), finally resultingin fibrin generation and clot formation.
On the other hand, excessive or pathological activation ofcoagulation is the common underlying cause of thromboemboliccardiovascular diseases, responsible for one in threedeaths. Anticoagulants, drugs that interfere with the coagulationsystem, are the cornerstone of the prevention and treatment ofvarious clinical manifestations of thrombosis, such as venousthromboembolism (VTE), atrial fibrillation (AF), andthrombosis on mechanical prosthetic heart valves.
The first generation of oral anticoagulants were discoveredin the United States in the 1930s as a result of the investigationinto the mysterious death of cows due to excessive bleeding.Dr. Karl Link was able to isolate the anticoagulant dicoumarol inmoldy clover used as cattle food.1 Warfarin, the first commerciallyavailable coumarin derivative, was first marketed as arodenticide. It was not until the 1950s that warfarin started tobe used in patients, gaining fame after being prescribed to USpresident Eisenhower after his heart attack in 1955. As the onlyavailable class of oral anticoagulants, vitamin K antagonists(VKA) became the standard for long-term anticoagulanttherapy and have been used by many millions of patients.
Although highly effective, the use of VKA is complicatedby its narrow therapeutic range and unpredictable pharmacokinetics.2,3 Due to genetic heterogeneity in VKA metabolism andnumerous interactions with food and drugs, treatment with VKArequires periodic monitoring of its anticoagulant effect throughthe international normalized ratio (INR).4,5 This monitoringimposes a significant burden on healthcare systems and patients,leading to an underuse of anticoagulants. Furthermore, evenwith laboratory monitoring, time in therapeutic range is oftensuboptimal, reducing the benefits of VKA treatment.
To overcome these challenges, a novel class of direct actingoral anticoagulants has been developed. These non-vitamin Koral anticoagulants (NOACs) or direct-acting oral anticoagulants(DOACs) feature more predictable pharmacodynamic andpharmacokinetic properties compared to VKA.6 Due to theirpredictable relation between drug dose and anticoagulanteffect, NOACs are designed to be used at a fixed dose, withoutroutine monitoring of coagulation parameters.
In this paper, we aim to give an overview of the pharmacodynamicand pharmacokinetic properties of the NOACs. Wewill discuss how the efficacy and safety of NOACs comparesto VKA for the treatment and prevention of VTE, as well asfor the prevention of stroke in patients with atrial fibrillation.Finally, we will discuss the practical use of NOACs in dailypractice
Four different NOACs are currently approved for VTEtreatment and for stroke prevention in AF patients. All 4NOACs target the common pathway of the coagulation cascadethat starts with the activation of clotting factor X, either throughthe extrinsic or the intrinsic coagulation pathway. Activatedfactor X (Xa) in turn activates prothrombin to thrombin, whichturns soluble fibrinogen into insoluble fibrin strands.
In contrast to VKA, which affects the maturation of differentclotting factors (II, VII, IX and X) (Figure 2A), NOACs bindto and inactivate a single activated clotting factor. Rivaroxaban,apixaban and edoxaban are direct factor Xa-inhibitors, whereasdabigatran is a direct thrombin inhibitor (Figure 2B).
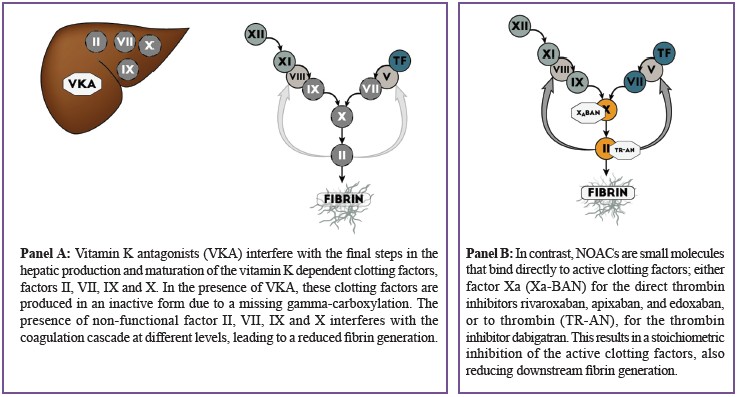
Figure 2: Comparison of mode of action of vitamin K antagonists (VKA) and Non-vitamin K antagonists Oral Anticoagulants (NOACs).
All NOACs are non-peptidic small molecules that share ahigh affinity (Ki in the nanomolar range) and specificity (littleeffect on other serine proteases) for their respective targets.Dabigatran specifically and reversibly binds to the active siteof both free and fibrin-bound thrombin7. In contrast with theindirect Xa-inhibitor fondaparinux, the direct Xa-inhibitorsbind to the active site of Xa, and inactivate both free Xa andXa-incoporated in the prothrombinase complex.8-11
Due to their direct activity on a single clotting factor, thereis a close correlation between the plasma concentrations andthe anticoagulant effect of these agents. Because of thispredictable pharmacodynamic profile, pharmacokinetic factorsare the main determinant of the net anticoagulant effect in agiven patient.
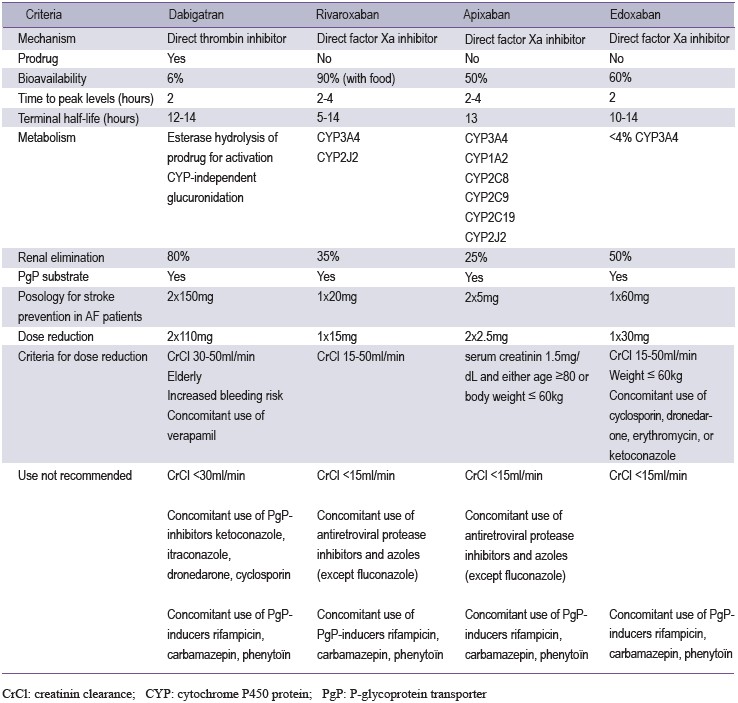
Table 1: Pharmacological properties of NOACs
Absorption and prodrug conversion
Dabigatran
To increase its bioavailability, dabigatran is formulatedas a prodrug, dabigatran etexilate. Overall, the bioavailabilityof dabigatran etexilate remains low (about 6%) and dependson an acid environment. Therefore, dabigatran capsulescontain pellets with tartaric acid. To ensure reliableabsorption, dabigatran capsules should not be opened orcrushed.12Peak plasma concentrations are reached within 2 hours oforal administration. During absorption, dabigatran etexilate israpidly converted in the active molecule dabigatran throughesterase-catalysed hydrolysis in the enterocytes, portal vein,and liver.13-15 Dabigatran has a relatively low plasma proteinbinding of approximately 35%, which allows the eliminationof dabigatran by hemodialysis.16
Xa inhibitors
Compared to dabigatran etexilate, the Xa-inhibitors do notrequire activation of a pro-drug, and have a higher bio-availability.For apixaban and edoxaban, bio-availability isindependent of food intake at about 50% (apixaban) or 60%(edoxaban), whereas the bio-availability of rivaroxabanincreases from 65% to > 90% when taken with food.17-19 Oralabsorption is rapid, and peak plasma levels are reachedwithin about 2 hours for edoxaban, and 2-4 hours forrivaroxaban and apixaban.6 Apixaban and rivaroxaban can becrushed and administered by nasogastric tube without relevantchanges in absorption.17,18 Since Xa-inhibitors have relativelyhigh plasma binding, dialysis is not very effective in reducingplasma levels.
Elimination
Dabigatran
The terminal half-life of dabigatran is around 12-14 hours,and is independent of the dose. About 80% of dabigatran isrenally excreted, with the remaining 20% of eliminationthrough conjugation and mainly biliar excretion.7 Because ofthe predominantly renal elimination, overall dabigatranexposure depends on the renal function. Patients with moderateor severe renal impairment have a 3- and 6-fold higher plasmaAUC of dabigatran compared to patients with normal kidneyfunction. In patients with a creatinine clearance below 30ml/min, half-life increases to over 24 hours16; therefore, dabigatranis not indicated in such patients.20 In elderly patients andpatients with a creatinine clearance of 30-50ml/min, a dosereduction of dabigatran is recommended. The elimination rateshould also be taken into account when interrupting dabigatrantreatment for elective invasive procedures or surgery. In patientswith normal renal function (clearance ≥ 80ml/min), it isrecommended to stop dabigatran 24 hours prior to mostprocedures, and 48 hours prior to high-risk procedures ormajor surgery.20 In patients with a clearance of 50-80ml/min,this should be increased to 1-2 days (standard procedure) or2-3 days (high-risk procedure), whereas patients with aclearance below 30ml/min require an interruption of 2-3 days(standard procedure) or 4 days (high-risk procedure) to allownormalization of hemostasis.20
Dabigatran is not metabolized by CYP isoenzymes, but isa substrate for the P-glycoprotein transporter (PgP).7 Therefore,PgP-inhibitors increase dabigatran plasma concentrations.Moderate hepatic impairment did not affect the rate of dabigatranglucoronidation. A study in patients with moderate hepaticimpairment (Child-Pugh B) showed similar pharmacokineticsand pharmacodynamics of single-dose administration ofdabigatran etexilate compared to matched controls with similarrenal function7. Nevertheless, the label restricts the use ofdabigatran in patients with hepatic enzymes of >2 times theupper limits of normal, and in patients with hepaticimpairment.12
Xa-inhibitors
All Xa-inhibitors are partially renally excreted, but to asomewhat variable extent. The contribution of renal eliminationin total drug clearance (elimination of unchanged drug) islower for apixaban (25%) compared to rivaroxaban (35%) andedoxaban (about 50%).9,17-19,21,22 The terminal half-life ofrivaroxaban is 5 to 9 hours in young subjects, and 11-14h inthe elderly.23 For apixaban and edoxaban, mean terminalhalf-lives are 13 and 10-14h, respectively.22 Although Xainhibitorsare less dependent on renal elimination comparedto dabigatran, renal impairment also increases the exposure toXa-inhibitors. Use of direct Xa-inhibitors is currently notrecommended in patients with a creatinine clearance below15ml/min, and should be used with caution in patients withmoderate renal impairment if other factors that may increaseexposure are present.20 Furthermore, patients with mild tomoderate reduction in renal function should be evaluated fordose reduction (see below).20
All Xa-inhibitors undergo hepatic metabolisation.Rivaroxaban is metabolised via CYP3A4, CYP2J2 and CYPindependentmechanisms.24 Apixaban is metabolised mainlyvia CYP3A4 and also to a lesser extent by CYP1A2, 2C8, 2C9,2C19 and 2J2.22,25,26 Edoxaban is approximately 50% hepaticallymetabolised, but mainly via CYP3A4-independentmechanisms (<4% CYP3A4).19 Thus, drugs that either induceor compete for CYP-metabolism may influence drug levels(see below). Furthermore, all Xa-inhibitors are substrates ofP-glycoprotein. Thus, co-administration of drugs with bothCYP3A4 and P-gp inhibition properties (e.g., ketoconazole,ritonavir) with Xa-inhibitors should be avoided as this mayreduce their elimination and significantly increase systemicexposure.20
Clinical use of NOACs in VTE
Primary prevention of VTE
The clinical development of all NOACs follows a similarpattern. Clinical trials in the prevention of VTE after majororthopaedic surgery, using a venogram to assess their efficacyin preventing mostly asymptomatic venous thrombosis, is awell-established clinical development model to validate theefficacy and safety of new anticoagulants. The approval ofNOACs for the prevention of VTE in orthopaedic patients haspreceded other indications; dabigatran, rivaroxaban, apixabanand edoxaban all are approved in some parts of the world forpreventing VTE after total knee or hip replacement.
Acute treatment of VTE
When trials have established the efficacy and safety of theNOAC in the prevention of VTE after major orthopaedicsurgery, large-scale trials are being initiated for the treatmentand secondary prevention of VTE, and for stroke preventionin patients with atrial fibrillation.The study design and results of the phase III trials withNOACs in VTE treatment are summarized in Table 2.
Table 2: Overview of phase III studies of NOACs in the acute treatment of VTE
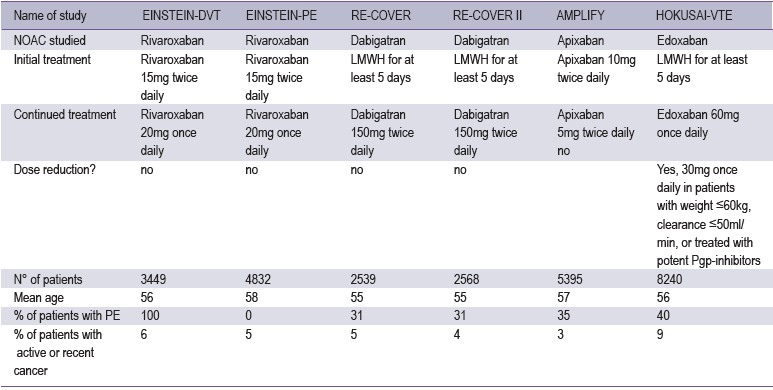
The EINSTEIN program investigated the efficacy andsafety of rivaroxaban for the treatment of acute DVT(EINSTEIN DVT) and acute PE (with or without symptomaticDVT, EINSTEIN PE), with a total of over 8000 patients widthVTE27. Rivaroxaban was consistently shown to be non-inferiorto standard enoxaparin/VKA therapy for the reduction ofrecurrent VTE in EINSTEIN DVT and EINSTEIN PE. In theDVT study, there was a trend for a superior efficacy outcomewith rivaroxaban compared with enoxaparin/VKA (2.1% vs3.0%, respectively; HR = 0.68; p = 0.08), which was not observedin the PE study (2.1% vs 1.8%; HR=1.12; p = 0.57)28. InEINSTEIN PE, a significant reduction in major bleeding by50% was observed in patients receiving rivaroxaban whencompared with those receiving enoxaparin/VKA (1.1% vs2.2%, respectively; HR = 0.49, p = 0.003), whereas EINSTEINDVT showed a 35% reduction in major bleeding with rivaroxabancompared to warfarin that did not reach statistical significance(0.8% vs 1.2%, respectively; HR = 0.65; p = 0.21)29.
The efficacy and safety of a short course of low molecularweight heparin (LMWH) followed by dabigatran has beeninvestigated for the treatment of acute symptomatic VTE inthe RE-COVER study program, including over 5,000patients30. Dabigatran showed non-inferiority in the reductionof recurrent VTE compared with warfarin, with a similarsafety profile, in a general VTE population, which includedpatients with DVT (~69%), PE (~21%) and both DVT and PE(~9%) across both treatment arms30. The RE-COVER II studyconfirmed non-inferiority of dabigatran.
The AMPLIFY program compared the use of apixabanwith warfarin in over 5,000 patients with VTE. Compared withwarfarin, apixaban was as effective in the prevention ofrecurrent VTE, but was associated with an almost 70%reduction in the risk of major bleeding events31.
In HOKUSAI VTE, the largest phase III VTE study,LMWH followed by edoxaban was as effective (HR 0.83,95%CI 0.60-1.14) as well-controlled warfarin in preventingrecurrent VTE, with a similar risk of major bleeding32.
For the clinician, it is important to understand how thedesign of the studies influences the initial treatment phase ofthe various NOACs. In the clinical studies with dabigatran andedoxaban, the initial treatment was therapeutic (LMW)heparin,whereas in the clinical trials with rivaroxaban and apixaban,a single drug approach has been investigated with an intensifiedNOAC regimen for 1 (apixaban) or 3 (rivaroxaban) weeks.Therefore, treatment with rivaroxaban or apixaban can bestarted with an all-oral regimen, whereas initial parenteralLMWH is required before initiating dabigatran or edoxaban.When starting an all-oral treatment, it is crucial to ensuring aproper switch from the higher initial dose to the lowermaintenance dose.
Taken together, these studies have included almost 30,000patients with acute symptomatic VTE. A meta-analysis of thestudies confirms that for the treatment of acute VTE, NOACsare as effective as well-controlled VKA, while offering a 40%reduction in major bleeding (HR 0.61, 95%CI 0.45-0.83)(Figure 3). NOACs are now the preferred treatment choice forthe majority of patients with acute VTE. Subgroup analysisshowed no difference in efficacy in patients with high vs lowbodyweight. In elderly patients and in patients with moderaterenal insufficiency, the relative benefit of NOACs was evenhigher due to a higher relative efficacy of NOACs comparedto warfarin33. In the approximately 1,500 patients with activeor recent cancer who were included in these studies, NOACswere more effective than and at least as safe as warfarin.Nevertheless, guidelines currently still recommend LMWH asa first-line treatment in cancer-related thrombosis, awaitingthe results of a head-to-head comparison of LMWH withNOACs. Results of the first of those studies, the HOKUSAICancerstudy, which compares the use of edoxaban withLMWH in patients with cancer-related VTE, are awaited atthe end of 2017.
Long-term secondary prevention of VTE
The efficacy of long-term treatment and secondary preventionof symptomatic VTE with dabigatran was demonstrated in theRE-MEDY and RE-SONATE studies (see Table 3)30,34.
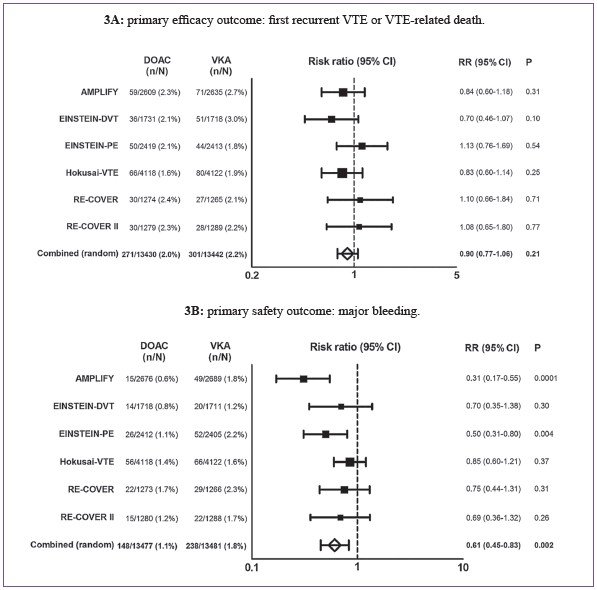
Figure 3: summary of results of phase III trials of NOACs in the treatment of acute VTE.
Table 3: Clinical trials of new oral anticoagulants and available alternatives in the extended treatment of secondary prevention of VTE:design and summary of results
Apixaban has been evaluated for the extended treatmentin patients who had completed 6 to 12 months of anticoagulationand for whom there was clinical equipoise to continue or tostop anticoagulant Both tested doses (2.5 mg and 5 mg twicedaily) were effective in preventing recurrent VTE, without asignificant increase in major bleeding complications.35
The EINSTEIN Choice compared low-dose aspirin withrivaroxaban in the ‘therapeutic’ dose (20mg once daily) or the‘prophylactic’ dose (10mg once daily) in patients whocompleted at least 6 months of anticoagulation after an acuteVTE event. Importantly, this study confirmed the importantresidual risk of VTE recurrence after stopping anticoagulanttherapy. Patients with unprovoked VTE had a risk of recurrenceof more than 5%/yr. after switching from anticoagulation tolow-dose aspirin. Both doses of rivaroxaban reduced the riskof recurrence by about 70%, without significantly increasingthe risk of major bleeding.36
Based on these studies, the use of a lower intensity ofanticoagulation seems an effective and safe strategy for thelong-term secondary prevention of venous thromboembolismin a large majority of patients. Evidently, a regular re-assessmentof the benefits and potential downsides of prolongedanticoagulation is required in patients receiving long-termanticoagulation, with special attention to modifiable riskfactors for bleeding.
The study design and results of the phase III trials with NOACs in the prevention of stroke and systemic embolism in patients with AF are summarized in Table 4.
Table 4: overview of phase III studies of NOACs in the prevention of stroke and systemic embolism in patients with non-valvular AF
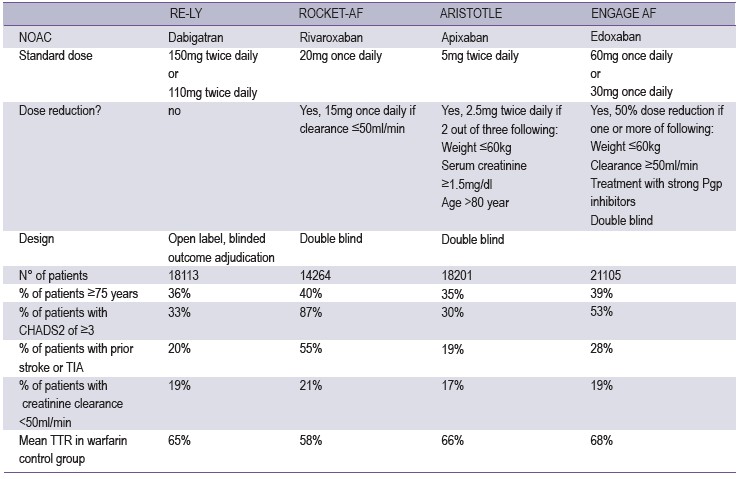
The landmark trials in the 1990s showed that anticoagulationbased on VKA targeted to an INR of 2-3 reduces the risk ofstroke in patients with AF by about 65 to 70%, while alsoreducing mortality.37,38 However, these studies wererelatively small. A new era of anticoagulation was started withthe publication of the RE-LY study, the first study of a NOAC,dabigatran, in patients with atrial fibrillation.
The RE-LY was an open-label study with blinded outcomeadjudication, randomizing patients with AF and at least onerisk factor for stroke to one of three arms: warfarin, dabigatran110mg twice daily, or dabigatran 150mg twice daily. In termsof efficacy, both doses of dabigatran were at least as effectiveas warfarin in preventing stroke and systemic embolism, withthe higher 150mg bid dose offering a significant reduction instrokes and ischemic events, as well as in ischemic strokesalone, on top of the well-known protective effect of warfarin.Whereas the long-standing concept was that more effectiveanticoagulation was always balanced with a higher risk ofbleeding, both doses of dabigatran were at least as safe, witha significant reduction of major bleeding for the lower (110mgbid) dose of dabigatran. Importantly, the most severe bleedingmanifestation of intracranial hemorrhage was strongly andsignificantly reduced by 70% by both doses of dabigatran.39
The ROCKET-AF study showed that a once-daily dose of 20 mg of rivaroxaban, reduced to 15 mg in patients with acreatinine clearance of below 50ml/min, lowered the risk ofthe combined endpoint of stroke and systemic embolismcompared to warfarin, without increasing the risk of majorbleeding. Importantly, this study included a high-risk population:the mean age was 73 years, and 87% of study participants hada CHADS2 score of 3 or more, with more than half of allpatients having a prior stroke or TIA.40
The ARISTOTLE trial compared warfarin with a twicedaily 5mg dose of apixaban, reduced to 2.5 mg twice daily ifat least two of the following criteria were met: age above 80, serum creatinine above 1.5mg/dL, and body weight ≤60kg. Thisregimen was associated with a 20% reduction of stroke andsystemic embolism, a 30% lower risk of major bleeding, and asignificant 10% reduction of mortality, compared to warfarin.41Finally, the ENGAGE-AF study randomized over 21,000patients to warfarin or two different doses of edoxaban: 60 or30mg twice daily. In each edoxaban arm, the dose was halvedin patients who had either a low body weight (≤60kg), acreatinine clearance of below 50ml/min, or who were treatedwith verapamil, quinidine, or dronedarone. Compared withwarfarin, edoxaban 60mg reduced both stroke/systemicembolism and major bleeding by about 20%. The lower, 30mgdose (reduced to 15 mg in patients with the above-mentionedrisk factors) was associated with an even larger reduction ofmajor bleeding of more than 50%, and was as effective aswarfarin to prevent the combination of stroke or systemicembolism. However, because of an excess of ischemic strokecompared with warfarin (40% increase), the 30mg dose (15mgin patients meeting dose reduction criteria) was not marketedfor stroke prevention.42
When combined in a meta-analysis, all NOACs show to beat least as effective to prevent stroke or systemic embolism, witha 20% reduction on top of the protection that VKAs had alreadyshown to offer. The reduction in strokes is mainly driven by adramatic decrease of the anticoagulation-associated risk inhemorrhagic strokes, reduced by more than 50%. (Figure 4)
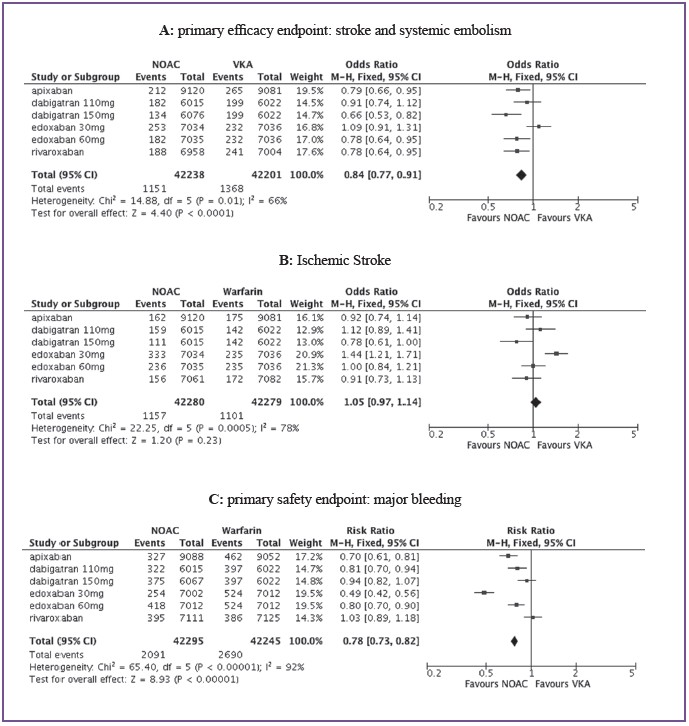
Figure 4: Overview of results of phase III studies of NOACs in the prevention of stroke and systemic embolism inpatients with non-valvular AF.
NOACs: less bleeding, but shift in bleeding patterns
When looking at overall bleeding, apixaban, both doses ofedoxaban, and the lower dose of dabigatran had significantlylower rates of major bleeding compared to warfarin, with astatistically neutral effect of dabigatran 150 mg and rivaroxaban.The magnitude of the reduction ranged from about 20% reduction for edoxaban 60mg and dabigatran 110 mg, to a morethan 50% reduction for the lower dose of edoxaban. In a pooledanalysis, major bleeds were reduced by 20% for DOACscompared to warfarin, with an absolute risk reduction of 0.72%per year.43 This translates into a NNT of 140, suggesting that for every 140 patients treated with DOACs as opposed towarfarin, one major bleeding episode is prevented (Figure 4C).
All DOACs significantly reduced the incidence of hemorrhagicstroke compared to warfarin, with reductions rangingfrom 41% (rivaroxaban) to 74% (dabigatran 150 mg) (Figure 5). In contrast, there was in increase in GI bleeding comparedto warfarin for rivaroxaban and for the higher doses ofdabigatran and edoxaban, but not for low-dose dabigatran andapixaban. The only DOAC regimen to reduce GI bleeding waslow dose edoxaban43 (Figure 5).
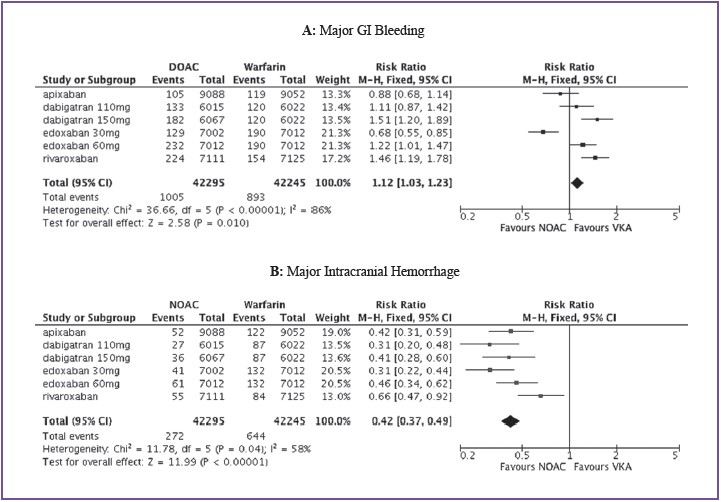
Figure 5: Differences in organ-specific bleeding patterns: major bleeding in GI tract (panel A) vs intracranial hemorrhage(panel B) of NOACs compared to warfarin.
Taken together, these data suggest a clear discrepancy in GIbleeding as compared to overall (major) bleeding, leading toeither a higher GI bleeding rate despite a neutral effect onoverall major bleeding (dabigatran 150 mg, rivaroxaban), areduction in major bleeding but no impact on GI bleeding(dabigatran 110 mg, and apixaban), or a reduction in majorbleeding with an increase in GI bleeding (edoxaban 60 mg). Onlyfor edoxaban 30mg, GI bleeding and major bleeding were bothreduced. One explanation of this finding may be that the directaction of the DOACs combined with high intra-luminal dosesdue to incomplete absorption lead to increased local anticoagulantactivity at the level of the gastro-intestinal wall.Besides causing fewer bleeds, NOAC-associated bleedingin patients from clinical trials has also been shown to have abetter outcome.
Who not to treat with a NOAC?
The large phase III trial programs, both in VTE (almost 30000 patients) and in AF (over 70 000 patients) have allowedto extensively study important patient subgroups. Althoughfrailer patients have a higher risk of both thrombo-emboliccomplications as for anticoagulant-related bleeding, overall,the relative benefit of NOACs is most pronounced in frailer patients, including patients over 75 years old, and patients withmoderately reduced renal function. Importantly, accumulating‘real-world evidence’ shows that the outcomes in patientsoutside clinical trials are very consistent with the findings fromthose studies, further reassuring clinicians about the favorablerisk-benefit profile of NOACs.43
However, some important patient groups were not studiedin those large clinical trials. Very few patients with a creatinineclearance of below 30ml/min were studied, as this was anexclusion criterium in most of the studies. As all NOACs arepartially renally excreted, drug levels are higher in patientswith lower creatinine clearance. This is especially true fordabigatran, which is almost exclusively excreted via the kidney(80% renal clearance). Thus, NOAC doses should be adjustedaccording to the label in patients with moderately reducedkidney function. With proper dose adjustment and carefulfollow-up of the renal function, NOACs can be used withcaution in patients with a creatinine clearance of down to 30ml/min, and down to 15 ml/min in selected patients for NOACswith relatively lower renal excretion (apixaban, edoxaban).However, NOACs are contra-indicated in patients withadvanced renal failure (creatinine clearance ≤ 15 ml/min) andin dialysis patients.20
NOACs cross the placental barrier in animal models, andtherefore are contra-indicated in pregnant women. Similarly,based on potential exposure, NOACs should not be used inlactating women.6,12,17-19
A single study evaluated the use of dabigatran in patientswith mechanical artificial heart valves. As this study showedmore thrombotic and more bleeding complications with thetreatment regimen studied, NOACs should not be used inpatients with mechanical heart valves.44,45 Importantly, patientswith moderate to severe valvular disease, including severemitral insufficiency and aortic stenosis, were included in thephase III trials. The effects of NOACs were not different inthose patients compared to the overall study results. Similarly,NOACs can be safely used in patients who underwent valvereplacement surgery with a bioprosthesis, or who had valverepair surgery.
Coagulation monitoring and measurement of anticoagulant effect
Due to the unpredictable pharmacokinetics and narrowtherapeutic range, VKA require periodic monitoring of theanticoagulant effect to guide dosing. In contrast, NOACs wheredeveloped to be used without routine laboratory monitoring.All phase III clinical trials compared the use of a fixed,unmonitored dose of NOAC with monitored VKA therapy. Asdiscussed elsewhere, these trials showed that the use of a fixedNOAC dosing regimen, adapted to patient characteristics butwithout any laboratory monitoring, was at least as effectiveand safe as well-controlled VKA therapy. Furthermore, therelative efficacy and safety of NOACs vs. VKA was consistentregardless of age, body weight, renal function, and frailty. Thus, for the vast majority of patients, measurement of theanticoagulant effect of NOACs is not required for treatment.
Although NOACs in general do not require monitoring,the measurement of NOAC-related anticoagulation may behelpful in some specific situations. First, in patients presentingwith bleeding or requiring urgent interventions, a morespecific evaluation of the anticoagulant effect can help to guidefurther treatment. Second, in patients with several factors thatmay influence NOAC exposure (age, body weight, renalfunction, and concomitant medication), assessment of NOAClevels could be useful to prevent excess exposure toanticoagulants, which may increase bleeding.
By interfering with the common pathway of anticoagulation(Xa-thrombin), NOACs can prolong clotting times and affectroutine coagulation testing. Nevertheless, the effect of differentNOACs is variable. Dabigatran prolongs the thrombin time(TT), escarin clotting time (ECT), and the aPTT. An ecarin-basedchromogenic assay and a calibrated diluted thrombin time(dTT) provide a highly reliable estimate of dabigatran levels,whereas the aPTT provides an approximative quantificationof dabigatran levels, but has limited sensitivity. Dabigatranhas little effect on the prothrombin time (PT).46,47
Rivaroxaban, in contrast, leads to a dose-dependentprolongation of the PT. However, the sensitivity and the degreeof correlation with rivaroxaban plasma levels depends on thereagent used. Rivaroxaban has little effect on the aPTT.48 Theeffects of apixaban and edoxaban on standard clottingassays are limited, and routine assays are not very helpful inqualitative or quantitative assessment of plasma levels of eitherdrug.49,50
For all Xa-inhibitors, measurement of anti-Xa-activitystrongly correlates with drug levels, and a calibratedanti-Xa-assay can be used to quantitatively measure druglevels.20
Stopping anticoagulant therapy and reversal of theanticoagulant effect of NOACs
One of the main advantages of NOACs over VKAs is theirshorter half-life. In those situations where interruption ofanticoagulation is required, simply stopping the administrationof the NOAC will result in normalization of the hemostaticsystem over the following 24 to 48 hours. Importantly,however, due to the renal clearance of NOACs, this timewindow is longer in patients with reduced kidney function.
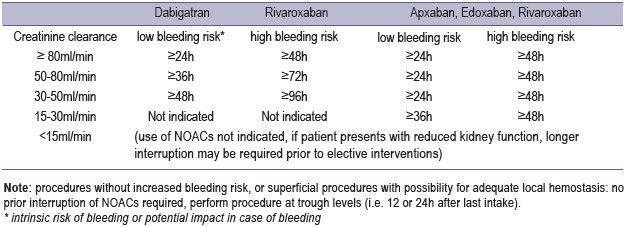
For elective interventions that require the interruption ofanticoagulation, current guidelines take into account the typeof NOAC, the kidney function, and the bleeding risk of theintervention (Table 5).
Table 5: Recommendations for interruption of NOAC therapy for elective surgical interventions: timing oflast intake of NOAC
In some situations, patients may require more rapidnormalization of their hemostatic system, for instance whenurgent surgery is required, or in case of major bleeding. Whenurgent reversal of anticoagulation is warranted, in vitro and invivo data suggest that prothrombin complex concentrates(PCCs), when given in high dose (50IU/kg), can normalizeclotting tests and can reduce blood loss and improve outcomein animal bleeding models.51
For dabigatran, a specific humanized monoclonal antibodyfragment that binds dabigatran with very high affinity has beendeveloped to specifically neutralize its anticoagulant effects.Idarucizumab, administered in a single 5g dose, immediatelyand completely neutralized the anticoagulant effect ofdabigatran, resulting in a direct and sustained normalizationof hemostasis in patients presenting with active bleeding orrequiring urgent surgery.52-54 Idarucizumab has been approvedfor dabigatran reversal, and is the first choice when urgentreversal of anticoagulation is required in patients treated withdabigatran.55 Since it specifically targets dabigatran, it has noeffect on the anticoagulant effect of VKAs, other NOACs, orheparines.
Andexanet alpha, an inactive factor Xa-analogue, is underdevelopment as a reversal agent for Xa-inhibitors, includingthe NOACs apixaban, rivaroxaban, and edoxaban, as well asthe indirect Xa-inhibitors such as heparine and LMWH. Abolus followed by a continuous infusion of andexanet alphawas shown to normalize clotting tests in volunteers treatedwith direct Xa-inhibitors and in patients presenting withbleeding.56,57 Regulatory approval is awaited in the upcomingyears, pending the results of an ongoing clinical study.Awaiting the availability of a specific reversal agent, PCCsshould be considered as hemostatic support in patients whorequire urgent reversal of direct Xa-inhibitor activity.
The development of oral anticoagulants with a more predictable pharmacokinetic and pharmacodynamics profile has allowed to provide reliable anticoagulation with a fixed once- or twice daily dose without need for drug level monitoring in millions of patients. The efficacy and safety of a fixed-dose NOAC is at least as good, if not better, than dose-adjusted VKA therapy over a wide range of patient’s subtypes for the prevention and treatment of VTE and for the prevention of stroke in patients with non-valvular AF. The large clinical study programs, ongoing new trials, and a large body of real-world evidence phase IV studies has increased our knowledge about anticoagulation in specific situations and patient populations, and has improved the quality of therapy with NOACs and VKA alike. Their ease of use, relative safety, and good efficacy has made the NOACs the drug of choice for many patients.









