Electronic ISSN 2287-0237
In 2014, ischemic heart disease (IHD) (also known as coronary heart disease), the most prevalent of the cardiovascular diseases, was the largest single leading cause of death in Thailand and worldwide.1 Currently, there are no effective treatments that simultaneously salvage the ischemic myocardium while also limiting the deleterious effects of reperfusion. Cardioprotection is a promising novel therapeutic approach for the treatment of IHD, as cardioprotective interventions have been shown to limit deleterious effects of ischemia reperfusion (I-R) injury by as much as 75%.2 However, its clinical translation has been limited due to its poor effcacy in the aged heart as evidenced by numerous experimental and clinical studies. The molecular basis of insensitivity to cardioprotective stimuli (i.e. by various pharmacological agonists) in the aged heart has been linked to specialized invaginations in the plasma membrane termed caveolae (little caves). Cardioprotective interventions such as ischemic preconditioning as well as numerous pharmacological approaches are reliant on the presence of caveolae which present a novel therapeutic target as enhanced expression of caveolae has been shown to be protective against a variety of stressors such as I-R injury in the young and aged heart.3 Besides its distinct morphology, caveolae can also be distinguished from other specialized membrane fractions (such as lipid rafts) due to the expression of the caveolin, cavin and Popdc (Popeye domaincontaining) family of scaffolding proteins.
The caveolin protein family consists of three members (Caveolin-1, -2 and -3) and the recently re-classifed family of cavins (Cavin-1/Ptrf, Cavin-2/Spdr, Cavin-3/Pkrcbp and Cavin-4/Murc). The most recent addition to caveolar coat proteins is the Popdc family which consists of three members (Popdc1/Bves, Popdc2 and Popdc3). Although caveolar bulbs also contain proteins such as Pacsin and EHD for caveolae function, they will not be discussed here due to spatial constraints (see review Han et al. 2016).4,5 As shown by Figure 1, caveolae house ion channels, cardioprotective receptors as well as their subunits, thereby providing spatial and temporal organization of cellular signaling events that are essential for stress resistance in the heart.6,7 Furthermore, cardioprotective methods such as ischemic preconditioning (IPC) and anesthetic preconditioning have been shown to increase cardiomyocyte caveolae formation and are associated with reduced infarct sizes and improved recovery in mice.8,9 Besides co-localizing numerous cardioprotective receptors such as β2-adrenoreceptors and effector molecules such as endothelial nitric oxide synthase (eNOS), a brief introduction of caveolins, cavins and Popdcs will be presented with emphasis on their roles as crucial determinants of stress adaptation in the heart.
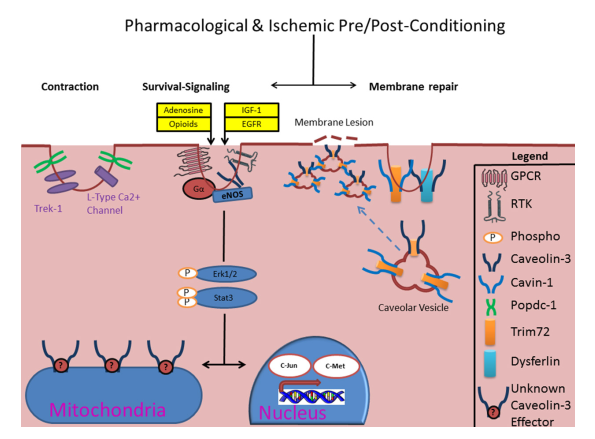
Figure 1: Schematic of caveolae-mediated protective signaling in the heart. It is hypothesized that IPC causes focal accumulation of adenosine and opioid substrates which causes autacoid stimulation of several GPCRs (i.e. Adenosine A1 & δ-opioid receptors) and receptor-tyrosine kinases (RTKs, i.e. IGF-1, EGFR, etc.), which are enriched in caveolae with their subunits, to initiate pro-survival signaling during stress which involves diverse 2nd messengers (i.e. Gα) and kinase signaling (such as Erk1/2) which converge on the mitochondria and/or nucleus. Notably, caveolae also house ion channels (i.e. L-type Ca2+) and may thus be a crucial determinant of post-ischemic recovery. Emerging body of evidence also suggests non-canonical roles for caveolins and cavins in regulation of membrane repair and mitochondrial integrity. Although Caveolin-3 is present in the mitochondrial membrane, it is not known whether it co-localizes with any mitochondrial proteins.
Caveolae can be distinguished from other members of the lipid raft family due to the expression of the caveolin family of scaffolding proteins. The caveolin protein family consists of three members, Caveolin-1 (Cav-1), Caveolin-2 (Cav-2) and Caveolin-3 (Cav-3). The resulting proteins are structurally related, with all three containing an identical stretch of eight amino acids (FEDVIAEP) within their N-terminus end. The signifcance of this structural/functional motif is unknown, although it is highly conserved across species and is therefore likely to have an important function.10Notable cardioprotective receptors that have been shown to co-localize in caveolae and caveolins include the δ-opioid, Bradykinin, A1 adenosine, and epidermal growth factor receptors.9,11-13 As shown by sucrose density gradients and confocal microscopy, caveolae have been shown to compartmentalize GPCRs (i.e. δ-opioid, A1 Adenosine receptor) and their subunits.7 For example, confocal and electron microscopy demonstrates co-localization of Cav-3 and Gαs within the intercalated disks of cardiomyocytes and in the sarcolemma.7 It should be noted that caveolae, in addition to enhancing signaling, are also capable of inhibiting signaling. The putative caveolin scaffolding domain (82-101) has been shown to inhibit many ligands that are cardioprotective including eNOS and matrix metalloproteinase-2 in the heart.14,15 All of these fndings suggest that caveolae in conjunction with caveolin proteins concentrate GPCRs and their subunits to allow effcient signal transduction.5
Cav-1 was the first caveolae marker protein to be discovered.16 Cav-1 displays 58% homology to Cav-2 and 85% to Cav-3.17 Cav-1 protein is ubiquitously expressed and is responsible for caveolae formation in non-muscle type cells such as endothelial, adipose and epithelial type tissue.18 Cav-1-deficient mice show a dramatic reduction (≈50%) in life span when compared to wild-type mice.19 Phenotypically, Cav-1-deficient mice show a combination of pulmonary fibrosis, pulmonary hypertension, and cardiac hypertrophy.19 As shown by electron micrograph images, double Cav-1/ Cav-3-deficient mice show that caveolae disappear in cardiomyocytes and in the adjacent endothelial cells while Cav-3- deficient mice show loss of caveolae in the cardiomyocyte but not in the adjacent endothelial cell.20 While Cav-1-deficient animals show disappearance of caveolae in the endothelial cells but not in cardiomyocytes, thus suggesting Cav-3 is responsible for sarcolemmal caveolae formation.20 Cav-1 has a pivotal role in stress resistance in the liver, brain and the heart as Cav-1-deficiency in these tissue result in increased sensitivity to ischemic injury.21-23 Conversely, enhanced expression of Cav-1 has been shown to be cytoprotective against ischemia-reperfusion injury in the brain and the heart.24,25 Consistent with these findings, following injurious stimuli, Cav-1-deficient hearts display increased cytokine/ chemokine such as IL-4 and TGF-β expression for up to 3 days, resulting in significant macrophage infiltration, fibrosis and adverse remodelling.26,27 Cav-1-based peptides (which have ≈5-10 hour half-life) present an attractive opportunity for the treatment of cardiovascular diseases such as acute myocardial infarction as Cav-1 peptide delivery in the vasculature and the heart has been shown to suppress acute inflammation vascular leakage and contractile dysfunction following ischemia-reperfusion.24-26
The mechanism of protection afforded by Cav-1 against I-R injury has been shown to involve its scaffolding domain which interacts with eNOS and tyrosine kinase Src-mediated phosphorylation of Cav-1.22,24 Protection afforded by Cav-1 peptide delivery is reliant on eNOS-mediated mechanisms as pharmacological inhibition (L-NAME) of eNOS has been shown to reverse this protection, resulting in increased vascular adherence, leukocyte infiltration and reduced recovery.24,25 Cardioprotective methods such as IPC and anesthetic preconditioning result in Src-mediated phosphorylation of Cav-1 resulting in cardioprotection.22 Although the significance of this phosphorylation is unknown in the heart, the protection afforded by these methods has also been shown to be reliant in phospho-Cav-1 as Src inhibition abolishes Cav-1 phosphorylation and cardiac protection associated with it.22
Although it has yet to be shown in cardiomyocytes, Cav-1 is crucial for mechanoprotection and stress resistance in several types of cells. In 3T3 fibroblasts phosphorylation of Cav-1 has been shown to be required for Dynamin1-mediated internalization of caveolae, with internalization of caveolae thought to be a crucial step in plasma membrane repair in T lymphocytes and in endothelial cells.28-31 Endothelial caveolae have been shown to protect cells from membrane damage during increases in cardiac output and hypoxia in vivo as Cav-1-defciency has been shown to result in decreased plasma membrane integrity following increased cardiac output and hypoxia.32 Though not widely promoted, preservation of endothelium should also be considered to be an important aspect of cardioprotective interventions as coronary microvascular dysfunction has been shown to correlate with reduced myocardial salvage, increased infarct size, adverse ventricular remodeling and clinical outcomes.33-35 Therefore, therapies that enhance Cav-1 expression may limit both microvascular dysfunction and ischemia-reperfusion injury and present a new therapeutic target. Despite the growing evidence that Cav-1 is a prerequisite for variety of cellular functions, the exact mechanisms of Cav-1-mediated cardioprotection remain largely uncharacterized, perhaps owing to the popularity of Cav-3. Whether Cav-2 has any significant impact on I-R resistance remains to be shown.
Cav-3 displays 85% similarity to Cav-1 and 60% similarity to Cav-2.17 Cav-3 expression is limited to muscle-type tissue with skeletal muscle and cardiomyocytes showing high expression of Cav-3.36 The importance of Cav-3 in these tissues can be highlighted by the existence of “caveolinopathies” with 30 known mutations in the Cav-3 gene, which results in a variety of clinical phenotypes.37 Cav-3 is expressed in cardiomyocytes and is responsible for caveolae formation in cardiomyocytes.20 Cav-3 protein expression is down-regulated with ischemiareperfusion whilst cardioprotective stimulus such as IPC is known to increase Cav-3 protein and caveolae formation.8,38 Cav-3-deficiency and cholesterol depletion (i.e. using Methly- β-cyclodextrin) which depletes caveolae, abrogates the cardioprotective benefits of IPC, non-G-protein-coupled-receptor (such as anesthetic preconditioning) and GPCR-induced preconditioning thus highlighting caveolae as important signaling centers.39-41 All of these findings are consistent with the evidence that caveolae in cardiomyocytes has been shown to host numerous G-protein-coupled receptors as well as their subunits.7 Collectively these studies suggest that various forms of cardioprotective stimuli are reliant on the presence Cav-3 and caveolae, with caveolae depletion via pharmacological approaches and Cav-3-defciency shown to ablate the cardioprotective effects of these stimuli.
Besides localizing cardioprotective receptors, Cav-3 also interacts and co-localizes with notable mediators of cardioprotection such as eNOS in the heart although it must be noted that this localization of eNOS is not exclusive to caveolae as eNOS is also present in non-caveolae fractions.42 Interaction of Cav-3 with eNOS is crucial for IPC-mediated cardioprotection as pharmacological disruption of caveolae via MβCD treatment prevents IPC-induced increase in protein S-nitrosylation (an important protective post-translational protein modification) as well as blocking phosphorylation of eNOS and Akt leading to impaired recovery and increased infarct sizes.43 Mitochondria have been shown to be a crucial target for protein S-nitrosylation as pharmacological caveolae depletion (via MβCD) results in reduced IPC-mediated protein-S nitrosylation of subsarcolemmal mitochondrial populations.43 Interestingly, Cav-3-overexpression does not change the expression and activity of NOS isoforms in the heart.8 Association of eNOS and iNOS with Cav-3 may be an important feature of sex-related differences in cardioprotection as association of eNOS and iNOS with Cav-3 is significantly higher in ischemia-reperfused females than in males.44
Ischemic stress has been shown to results in multiple tiny breaks in the plasma membrane that, without immediate repair results in irreversible cell injury leading to necrosis.45 Consistent with their roles as protective elements and sensors in the plasma membrane, caveolae and caveolins are also required for plasma membrane repair mechanisms in cardiomyocytes as well as other cell types. To support this, knockdown of Cav-3 diminishes plasma membrane repair capacity following membrane injury resulting in enhanced cell death whilst IPC relocates dynamin from the cytosol to caveolae which may assist caveolae in plasma membrane repair.29,46 Cav-1 and Cav-3-defcient cells show abnormal Dysferlin trafficking (involved in membrane stability), similar to aberrant Dysferlin localization in caveolinopathies which suggest that caveolins may be important in targeting Dysferlin to the membrane for sarcolemmal stability.47,48 Similarly, mutations in Cav-3 result in retention of plasma membrane repair moieties DysferlinTrim72 in the Golgi apparatus resulting in defective membrane repair.49 The presence of cholesterol (which is also enriched in caveolae) in the plasma membrane has been shown to be required for proper targeting of these proteins as pharmacological disruption of cholesterol impairs the plasma membrane repair response and the targeting of caveolae coat proteins to the plasma membrane.50,51 More recently, Cav-3 has also been shown to co-localize with autophagy markers such as beclin-1 and light chain 1 which are reduced in Cav-3 knockdown cells and associated with reduced adaptation to cardiac stress.52
Recently, a new member of caveolae proteins termed cavin (Cavin-1, -2, -3 and -4) has been re-classifed for their roles in caveolae biogenesis and localization. These four cavin proteins share varying degrees of homology relative to each other, Cavin-1 is 66% similar to Cavin-2 and 59% similar for both Cavin-3 and Cavin-4.53 All cavin proteins display two PEST (proline, glutamic acid, serine and threonine-rich domains) as a signature motif, it is not known what function PEST domains have in the cavin proteins.54 It is known that PEST domains can promote protein degradation through µ-calpain and 26S proteasome system.55,56 All cavin proteins also share a conserved coiled-coil domain, leucine zipper and a membrane association domain.57,58 The coiled-coil domain of Cavin-4 has been shown to be required for sarcolemmal targeting of Cav-3 as mutations in this domain results in reduced Cav-3 protein and targeting to the membrane.59
Cavin-1 was initially described as a Polymerase I and transcript release factor (PTRF) as the dissociation of transcripts from Polymerase I and its tertiary structure required the presence of this protein.60 It was later renamed as a caveolae-related protein (cavin/cav-p60) as it was found to be particularly enriched in plasma membrane fractions and electron micrographs suggested distinct caveolae localization.61 Cavin-1 is moderately expressed in the heart with adipose and lung tissue also showing notable expression of Cavin-1.36 In the heart, Cavin-1 is enriched in sucrose density fractions 4-12 whilst Cav-3 is enriched in fractions 11-12 suggesting there may be distinct subcomplexes.8,62 However, IPC causes increased translocation of Cav-3 to fractions 4-5 which are particularly enriched in Cavin-1 protein.8,62 Cavin-1 localizes with Cav-3 and is essential for caveolae formation in some cell types while also simultaneously acting as an accessory protein.36,62 Cavin-1-defciency results in the loss of caveolae in a variety of cell types including lung epithelium, intestinal smooth muscle, endothelial cells, skeletal muscle and cardiomyocytes.36,63 Cavin-1 has been shown to be involved in the stability of Cav-1 and Cav-3 as knockdown of Cavin-1 causes caveolins to diffuse freely in the plasma membranes which eventually become internalized into the endolysosomal system for degradation.64 Rats with heart failure show significant Cavin-1 down-regulation accompanied by Cav-1 and Cav-3 down-regulation, consistent with suggestions that Cavin-1 stabilize caveolins.65
In experimental models, loss of Cavin-1 via mutations or knockout approaches show a phenotype of generalized lipodystrophy, glucose intolerance, cardio and muscular dystrophy.36,66 Consistent with these experimental observations, clinical mutations in the Cavin-1 gene has been shown to be associated with patients who displayed lipodystrophy, muscular dystrophy, cardiac arrhythmias and long-QT syndrome.63,67 Though its contribution/importance in canonical cardioprotective signaling is currently unknown, Cavin-1 is important in targeting plasma membrane repair machinery to sites of injury. Cavin-1 co-localizes with Trim72 (a.k.a. Mg53), which has been shown to be critical for membrane repair following I-R, leading to cardioprotection.68,69 The role of Cavin-1 in this repair mechanism suggests that Cavin-1 helps transport and bind Trim72 to cholesterol-rich regions (which may be caveolae) thereby anchoring the Trim72 molecule to enable membrane repair.68 This repair response was shown to be reliant on the presence of Cavin-1 and lipid rafts/caveolae as cholesterol depletion via MβCD disrupts the repair response.50,68 Cavin-1 knockdown also leads to a loss of Trim72-mediated responses following membrane damage (streptolysin O), and enhances cellular LDH release and FM1- 43 dye entry, further implicating caveolae involvement in mediating the plasma membrane repair response.68The role of Cavin-2, Cavin-3 and Cavin-4 in ischemia-reperfusion injury are largely uncharacterized. However, it is known that the membrane trafficking of Cav-3 involves the coiled-coiled domain of Cavin-4 as mutations in this domain result in retention of Cav-3 in the cytoplasm and reduces its protein (but not mRNA expression) and may therefore assist in normal caveolar functioning.59
Although not widely associated with caveolae, members of the Popeye domain-containing (Popdc) gene family have also been shown to be crucial for caveolae formation and caveolae-related functions.70 The Popdc family consists of three members: Popdc1, Popdc2 and Popdc3. Popdc2 and Popdc3 display 50% sequence identity on the protein level, while Popdc1 is only 25% similar to Popdc2 and Popdc3.71 Popdcs are ≈43-kDa transmembrane proteins that contain the two highly conserved sequence motifs (FL/IDSPEW/F and FQVT/SL/I) crucial for cAMP binding.72 The amino terminus of Popdc can be N-glycosylated, which is responsible for the apparent molecular mass of 58-kDa of Popdc1 on PAGE gels although it has a predicted mass of 43-kDa.71 Expression of Popdcs is restricted to muscle-type cells although some epithelial-type cells also express Popdcs such as those found in the central and autonomic nervous system and in epithelial cells of the epidermis, gastrointestinal tract, retina, lens, and cornea.73 There is differential expression of Popdcs within the heart; Popdc1 expression in the ventricular myocardium is lower than of the atria, while Popdc2 is homogenously expressed in both chambers.73 Currently, very little is known about the exact physiological function of Popdcs. They have been implicated in variety of processes such as chronotropic response, cell regeneration, regulation of membrane processes and ischemic tolerance.70,74,75
In the heart Popdcs are thought to maintain cardiac pace making as Popdc1 and Popdc2 disruption results in stress-induced sinus node dysfunction, resulting in chronotropic incompetence and long sinus pauses.74 More specifcally, in 8-month old Popdc1- and Popdc2-deficient animals display maladaptive cardiac pace making and age-related bradycardia in response to stress (swimming/exercise) when compared to age-matched wild-types.74 It is thought that the arrhythmias associated with Popdc disruption is due to loss of Trek-1 potassium channel activity, which co-localizes with Popdc1 and Popdc2.74 Indeed, Popdc1 has been identifed as a hub gene suspected in the initiation or progression of atrial fbrillation.78 These fndings are also consistent with the observations that caveolae house numerous ion channels related to contractile function and T-tubule organization.79,80 Clinical Popdc mutations have also been identifed which display similar phenotypes encountered with Cav-3 and Cavin-1 mutations. More specifcally, four novel mutations, which increase its transcriptional activity, have been identified for Popdc1 in patients with Tetralogy of Fallot while a missense mutation variant has been identifed in a family of 4 with cardiac arrhythmia and limb-girdle muscular dystrophy.76,77 Popdc1-defciency in the mouse does not result in abnormal morphology of the heart and the abnormal morphology observed with Tetralogy of Fallot may be due to its increased transcriptional activity and/or other genes that are perturbed with this condition.78
Emerging evidence suggests that Popdc1 is a crucial determinant of ischemic tolerance in the heart. Popdc1- deficient cardiomyocytes show significant reduction in caveolae with remaining caveolae being larger in size.70 Similar to Cav-3-defciency, Popdc1-defcient hearts show a loss of endogenous ischemic tolerance, as well as loss of IPC and anesthetic preconditioning (isoflurane) leading to enhanced infarct size and impaired recovery following I-R.40,41,70 Consistent with the role of caveolae in stress adaptation, both Popdc1 and Cav-3 appear to be down-regulated both in murine and human end-stage heart failure.81,82 Combined reduction of these crucial caveolar proteins in the hearts may contribute to the pathogenesis of heart failure. It is interesting to speculate whether the down-regulation of Popdc1 observed with I-R and heart failure can affect the activation of cardiac satellite/ progenitor cells, as activation and regeneration following injury in skeletal muscle models require Popdc1, although this remains to be shown for cardiomyocytes.71 To date, the importance of Popd2 and Popdc3 in caveolar biogenesis and their cellular roles is largely uncharacterized. Given their homology to each other is only 25%, the role of Popdc3 may be substantially different to that of Popdc1. Popdc2-defcency in zebrafish is associated with atrioventricular block and muscular dystrophy.83 Knockdown of Popdc3 in zebrafish does not result in ventricular repolarization defects whilst Popdc1 knockdown results in shortening of the zebrafish ventricular action potential duration.84 Reduced Popdc3 expression has been correlated with high risk and poor survival in patients with gastric cancer and may therefore be an important determinant of cellular signaling.
Caveolae are gaining increasing recognition in relation to cardiac aging as the expression of Cav-3 protein and formation of morphological caveolae have been shown to be reduced in aged mice myocardium (74-week old) as well as in the failing human heart.82,86,87 Similarly, age-related reductions in caveolins and caveolae expression are also reported in bladder smooth muscle,88 neurons89 and muscle cells.90 Due to the limited availability of clinical atrial/ventricular tissue, a detailed expression analysis of caveolae and its coat proteins has not been conducted in human specimens although the reduction of efficacy of GPCR-mediated cardioprotection has been documented in 75±2 year old human atrial preperations.86
In experimental models, the aged heart exhibits a stressintolerant phenotype with reduced sensitivity to various GPCR receptors, including A1AR, Bradykinin, and δ1-opioid receptors, which reduce I-R injury in young hearts but not in aged hearts.86,91 Concomitant with the loss of endogenous and GPCR-mediated protection is an age-related reduction of Cav-3 as shown for the mouse myocardium.86 As many cardioprotective receptors and their signaling subunits are co-localized in caveolae, a reduction in caveolae may play a key role in endogenous protective signaling and insensitivity to protective stimuli in the aged heart. In support of this, both Cav-3 and Popdc1-defcient hearts as well as pharmacological disruption of caveolae results in loss of endogenous ischemic tolerance as well as loss of IPC and pharmacological preconditioning.40,41,70,93
Restoration of caveolae in aged hearts represents an unexplored frontier of cardioprotective therapeutics as there is scarcity of studies that have evaluated the combined effect of restoration of caveolae and activation of cardioprotective stimulus in these hearts. To date, only two studies based on poster session abstracts report that cardiac-specific overexpression of Cav-3 leads to improvement in recovery of the aged mouse heart, which results in 60–65% recovery of pressure development and end diastolic pressure reportedly in the normal range.3,95 Furthermore, Cav-3-overexpression inhibits (via ANP) cardiac hypertrophy and fibrosis in response to aortic constriction, with enhanced survival rates (vs. wild-types), further supporting the involvement of Cav-3 and caveolae in stress-resistance in the heart.94 However, it has yet to be shown whether Cav-3-overexpression can restore the efficacy of GPCR and IPC-mediated cardioprotective signaling and whether the protection observed is comparable to that of young hearts. Thus further experiments measuring the efficacy of cardioprotective methods in the Cav-3-overexpressed myocardium are necessary and should be an area of future investigation as it has high clinical relevance. Taken together, age-related reduction of caveolar coat proteins may be responsible for the disruption of endogenous protective mechanisms as well as refractoriness to protective stimulus. Future studies should characterize the expression of individual members of caveolins, cavins and Popdcs in the aging murine and human myocardium as they remain poorly characterized with aging.
Caveolae present an attractive opportunity for the clinical translation of currently used experimental methods such as ischemic pre/post-conditioning and pharmacological methods (both GPCR-based & anesthetic) which are dependent on the presence of these microdomains.96 Although numerous studies have clearly indicated a pro-survival role for caveolae and its coat proteins in the young heart, little to no studies have opted to evaluate whether the cardioprotective method being tested is equally efficacious in both young and aged hearts. Despite their crucial roles in myocardial stress resistance, caveolae have not been targets of any clinical trials owing to the “disconnection paradigm” that has become inherent in clinical cardioprotection research.97-99 Therefore, caveolae represent an important opportunity to re-vitalize interest in translation of current cardioprotective methods as there is dwindling hope that any cardioprotective intervention will produce clinically viable results.98 To accomplish this, it has been suggested that treatments aiming to restore ischemic tolerance in the aged heart must first recreate the environment observed in the young hearts whilst addressing the deficit (i.e. reduced caveolae formation) created in the aged heart.97
One method that would address both of these issues is the use of adenoviral vectors which are relatively safe and currently used in clinical gene therapy trials.100 Indirect intracoronary injection of Cav-3 adenoviral vectors in Cav-3-defcient hearts has been shown to restore caveolae formation seven days after gene transfer and may thus be a suitable method to restore caveolae expression in the aged heart.101 Other clinically feasible methods that should be further evaluated is the administration of caveolin peptides as delivery of Cav-1 and Cav-3 peptides has been shown to reduce I-R/ hypoxia injury.24,102 It remains to be shown whether administration of Cavin-1 and Popdc peptides following I-R can achieve similar outcomes. Notably, Cav-1 peptide delivery has also been shown to display atherosclerotic lesions in the aorta by 42% and aortic sinuses by 21% should also be evaluated as an experimental prophylactic for the treatment of IHD.103 Recently, Cavin-4-defcient mouse hearts has been shown to exhibit signifcantly smaller infarct size and preserved cardiac function with Cavin-4 knockdown using small interfering RNA shown to reduce H2O2-induced cell death.104 Similarly, Cavin-2 knockdown has been shown to render cardiomyocytes resistant to hypoxia and H202 via Akt-mediated mechanisms.105 Thus Cavin-2 and Cavin-4 may also be novel therapeutic targets for limiting I-R injury in the heart.
Although surgical and pharmacological approaches have attempted to limit I-R injury (whether accidental or surgical), IHD remains the largest single leading cause of death in Thailand and worldwide. By co-localizing a variety of ion channels, plasma-membrane repair-related proteins and cardioprotective receptors and their subunits, caveolae have been shown to be crucial mediators of cardioprotective signaling as summarized in Figure 1. The contribution of Cav-3 in cardioprotection is relatively well characterized (and to lesser extent Cav-1). The recently re-classifed cavin and the Popdc family members are also beginning to contest and solidify their roles in stress resistance in the heart as members of the Popdc and cavin family of genes have also shown to be a crucial determinant of myocardial caveolae formation and cardio protective signaling. Although the age-related reduction in Cav-3 is documented in a variety of aged tissues, to this date the age-dependent expression of other caveolins proteins (Cav-1 & Cav-2), cavins and Podpc, (which are also important for caveolar functions), remain uncharacterized with cardiac aging and should be an area of further investigation. Increasing cardiac expression of Cav-3 may be a novel therapeutic approach for ischemic heart diseases as Cav-3 overexpression in aged hearts as this has been shown to reduce mitochondrial dysfunction, infarct size and improve recovery following I-R.3,8,95 Though yet to be shown, such an approach may also have the potential benefit of salvaging the existing spectrum of cardioprotective strategies and should be an area of future investigation.
We thank Dr. Rita Juneja for her assistance in manuscript preparation.









